

A case report on primary cutaneous anaplastic large cell lymphoma
Win Myat Oo1*, Sein Win2 and Nyein Chan Aung3
1. Department of Pathology, No (1) Defence Services General Hospital, Mingalardon, Yangon, Myanmar
2. Department of Clinical Haematology, Yangon General Hospital, Yangon, Myanmar
3. Department of Pathology, Defence Service Medical Academy, Yangon, Myanmar
Correspondence to: Dr Win Myat Oo, Department of Pathology, No (1) Defence Services General Hospital, Pyay Street, Mingalardon, Yangon 11021 Myanmar. Telephone: +95 940 930 7131 Email: winmo27@gmail.com
Conflict of interest: The authors declare that they have no conflicts of interest with the contents of this article.
|
Submitted: |
24 March 2021 |
|
Accepted: |
5 April 2021 |
|
Published: |
1 October 2021 |
Abstract
Primary cutaneous anaplastic large cell lymphoma (PCALCL) is a rare T-cell lymphoma, characterised by anaplastic large cells that are usually ALK negative but have high expression of CD30 and presents as a solitary or grouped nodules. It is the second most common type of cutaneous T-cell lymphoma, usually skin-limited disease and most frequently affects the trunk, face and extremities. In about 10% of the patients, it involves the regional lymph nodes as extracutaneous dissemination. We presented a 52-year-old male with ulcerated skin tumour mass at dorsal aspect of left hand near the base of little thumb and one regional lymph node enlargement in left bicep muscle. Skin tumour mass showed diffuse lymphomatous infiltrate in the dermal portion containing cohesive sheets of neoplastic medium to large cells mixed with few inflammatory cells and histiocytes. There was no epidermotrophism. Tumour cells were positive for CD3, CD30, MUM1 and high Ki67 expression. Heterogenous expression of CD4 and CD8 were noted. They were negative for CD20, CD5, EMA, TIA1, ALK and EBV/LMP1 expression and it matched with primary cutaneous anaplastic large cell lymphoma. Regional lymph node shows the same morphology and the neoplastic large cells had strong expression of CD3 and CD30.
Keywords: CD30 expression; immunophenotyping of lymphoma; primary cutaneous anaplastic large cell lymphoma; T-cell lymphoma
Introduction
Primary cutaneous anaplastic large cell lymphoma (PCALCL) is a rare T-cell lymphoma, characterised by anaplastic large cells that are usually ALK negative but have high expression of CD30 and presents as a solitary or grouped nodules(1). It is the second most common type of cutaneous T-cell lymphoma, usually skin-limited disease and most frequently affects the trunk, face and extremities. In about 10% of the patients, it involves the regional lymph nodes as extracutaneous dissemination(2). Histologically, the tumour lesions show a diffuse infiltrate containing large-sized T lymphocytes with the morphology of anaplastic cells with oval, round, or irregular nuclei, prominent eosinophilic nucleoli and abundant cytoplasm, but there is no epidermotrophism(3). PCALCL has no expression of anaplastic lymphoma kinase (ALK) and does not have gene rearrangements involving the ALK gene(4). The main differential diagnosis of PCALCL is lymphomatoid papulosis (LyP) because their morphologic and immunophenotypic features overlap significantly and as no biomarker to date has been found to reliably distinguish these entities. It is essential to correlate the pathologic findings with the clinical history. The clinical behaviour of LyP is characterised by recurrent and regressing crops of papules and nodules that aids the distinction from PCALCL(5).
Case Report
We presented a 52-year-old male with ulcerated skin tumour mass (2.0 x 2.0 x 0.8 cm) at dorsal aspect of left hand near the base of little thumb (Figure 1A) and one regional lymph node enlargement in left bicep muscle. The patient noticed the small nodule underneath the left little thumb of the left hand. It was gradually increased within three months and became ulcerated on the surface. During the clinical course at three months, he noticed that there was enlargement of lymph node in the left bicep muscle. The patient denied presence of fever, weight loss or night sweats accompanying his ulcerated mass. Surgical excision of tumour mass and removal of lymph node were done due to suspicious nature of this ulcerating mass and the first pathological report was non-Hodgkin lymphoma (NHL) infiltration in skin and lymph node.
He subsequently presented to the Department of Clinical Haematology, Yangon General Hospital for further assessment and management. H&E slides were reviewed, and skin tumour mass showed diffuse lymphomatous infiltrate in the dermal portion containing cohesive sheets of neoplastic medium to large cells mixed with few inflammatory cells and histiocytes (Figure 2A). There was no epidermotropism. Anaplastic large tumour cells had oval, round or irregular nuclei, prominent eosinophilic nucleoli and abundant cytoplasm (Figure 2B). Reactive lymphocytes were present in the edge of ulcer and periphery.
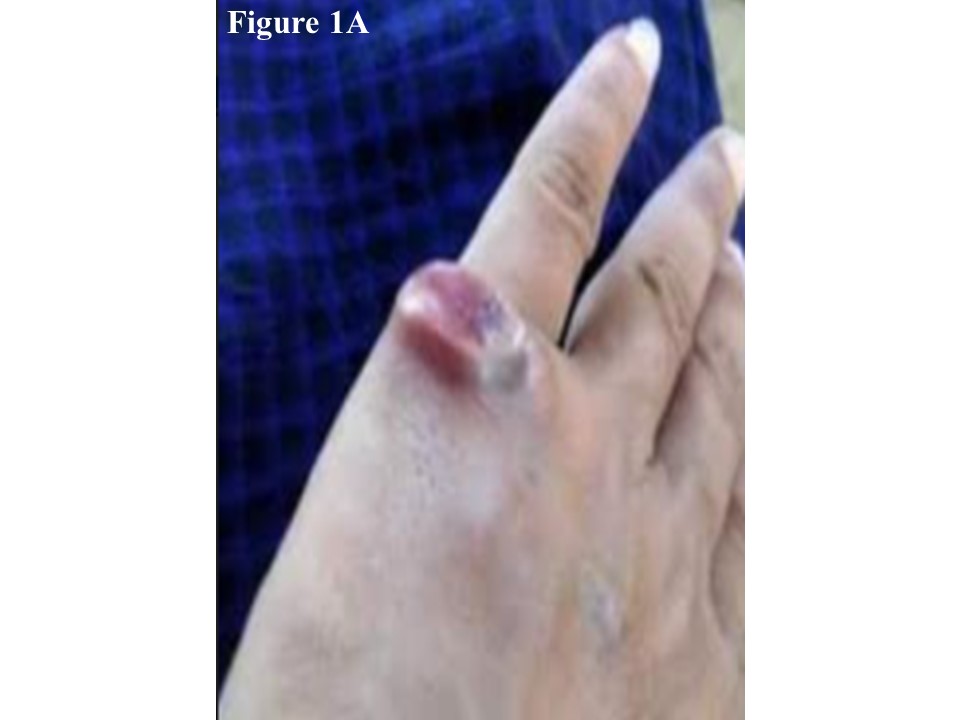

Figure 1 A 2.0 x 2.0 x 1.0 cm of ulcerated mass was developed at the base of little finger of left hand (A). Healed lesion was present after 4th cycle of chemotherapy (B).
Based on morphology, immunohistochemical stainings of CD3, C20, CD30, EMA, CD4, CD5, CD8, ALK, TIA1, MUM1, Ki67 and EBV/LMP1 were done. Tumour cells were strongly positive for CD3 and CD30 (Figures 2C and 2D). They were also positive for MUM1 and High Ki67 expression. Heterogenous expression of CD4 and CD8 were noted. They were negative for CD20, CD5, EMA, TIA1, ALK and EBV/LMP1 and it matched with primary cutaneous anaplastic large cell lymphoma. Regional lymph node showed the same morphology and the large neoplastic cells had strong expression of CD3 and CD30 (Figure 3).
After the surgical excision and four cycles of chemotherapy (CHOP), the lesion became cured (Figure 1B).
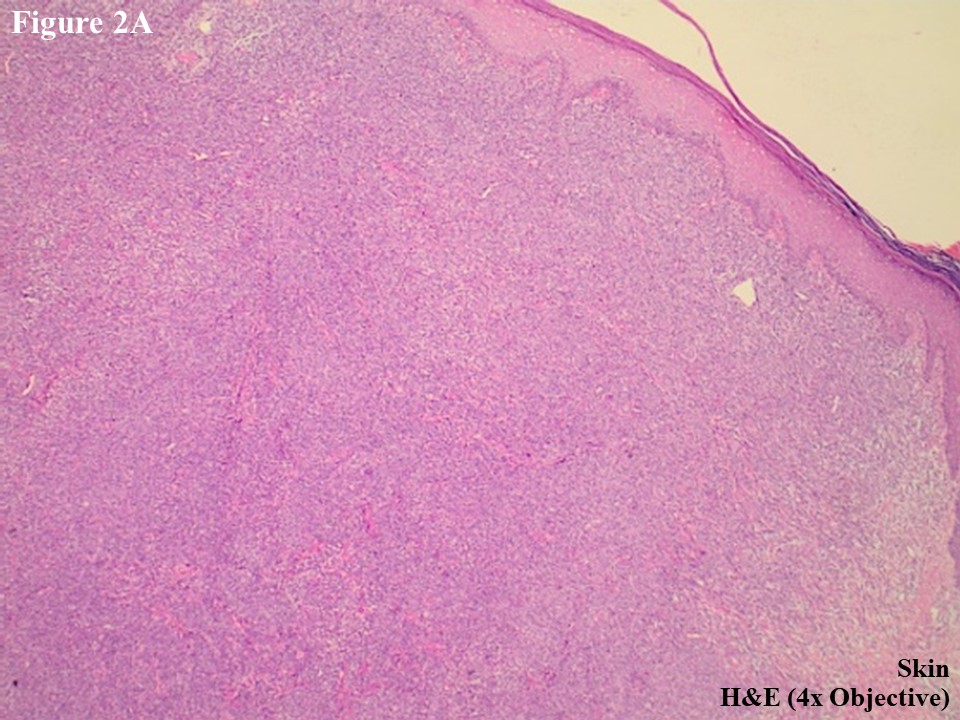
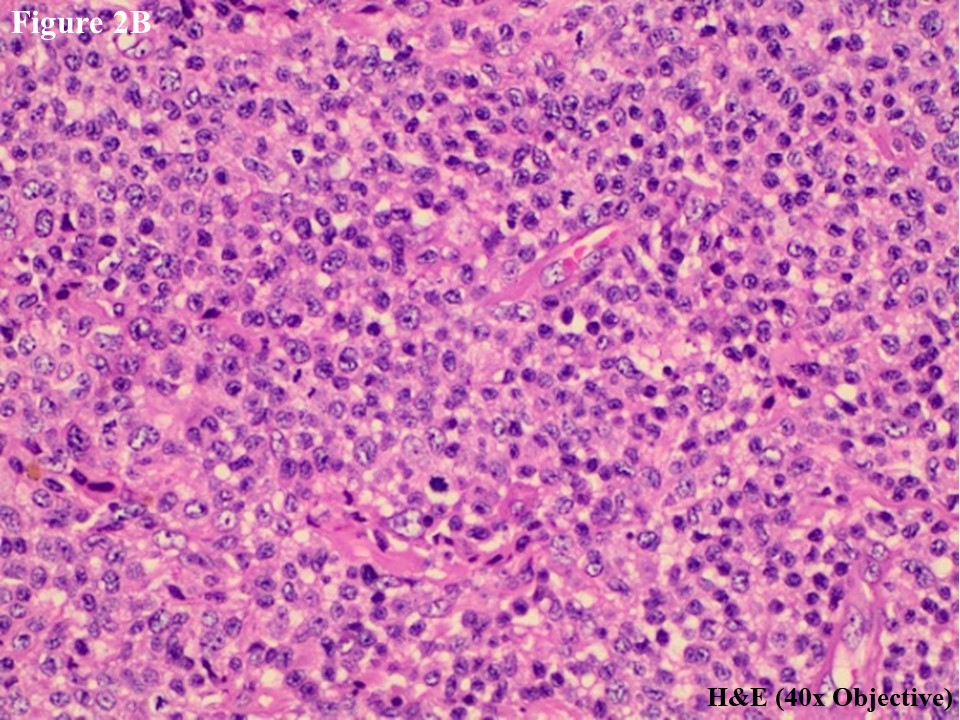

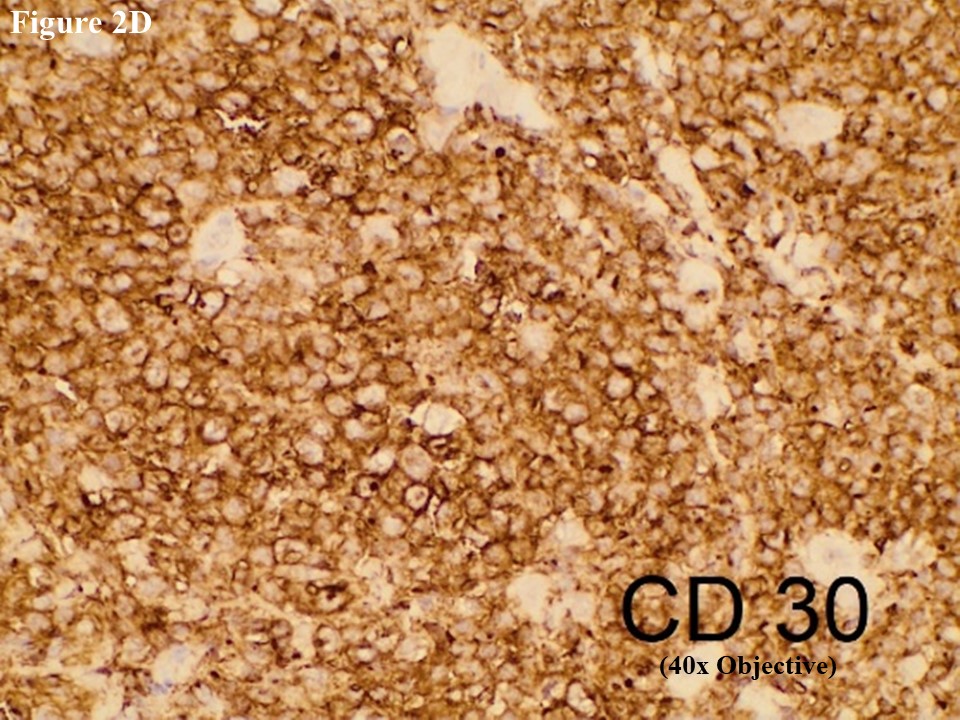
Figure 2 PCALCL. The skin tissue microscopically revealed cohesive sheet of medium- to large-sized neoplastic lymphoid cells in the dermis without features of epidermotrophism (A). The infiltrating anaplastic tumour cells had abundant cytoplasm with oval, round, or irregular nuclei and prominent eosinophilic nucleoli (B). These large tumour cells yielded positive immunoexpression of CD3 (C) and CD30 (D).
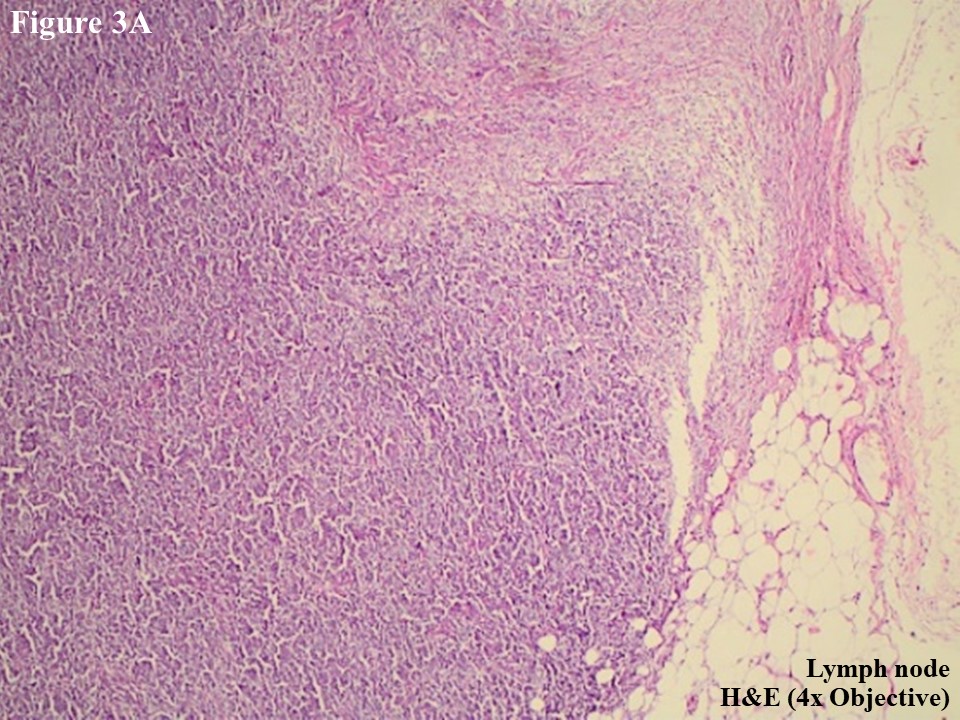

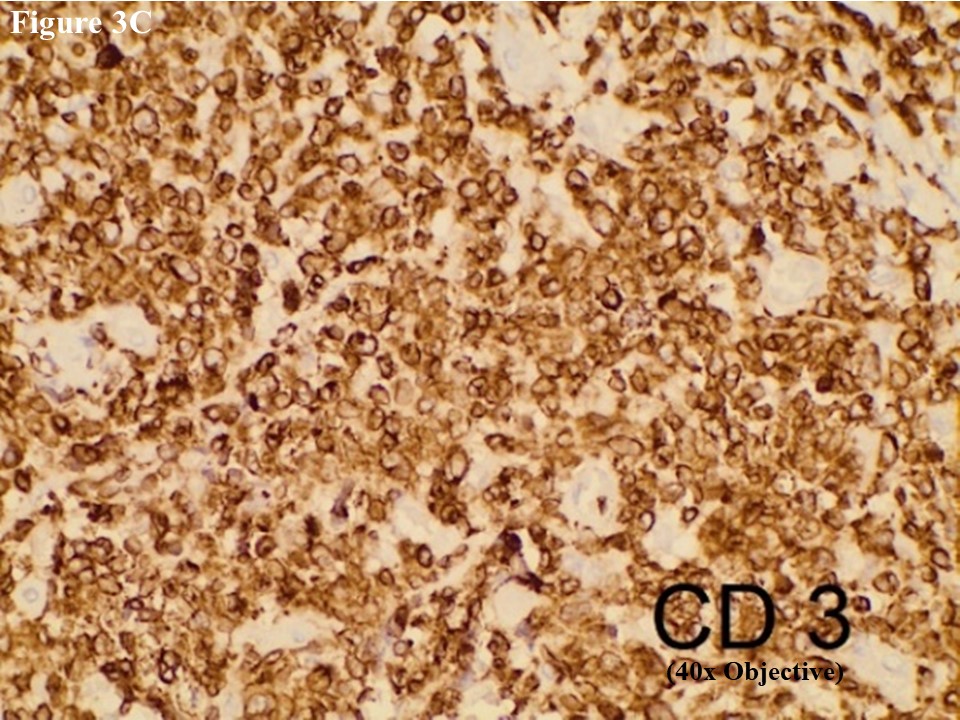
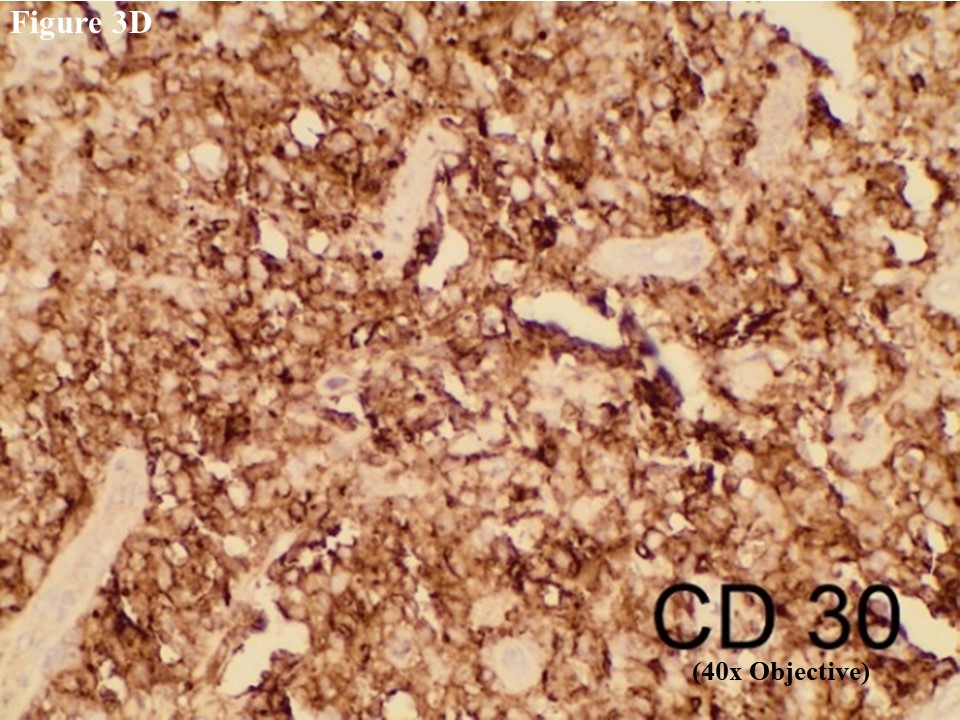
Figure 3 Regional lymph node involvement by PCALCL. Lymphoid architecture was totally distorted and was involved with diffuse cohesive sheet of anaplastic large cells (A). The anaplastic large cells had moderate amount of eosinophilic cytoplasm with oval, round or irregular nuclei (B). These large tumour cells yielded positive immunoexpression of CD3 (C) and CD30 (D).
Discussion
According to the WHO 2017 revised 4th edition, primary cutaneous anaplastic large cell lymphoma (PCALCL) is the second most common type of cutaneous T-cell lymphoma, the median patient age is 60 years and male to female ratio is 2 – 3 : 1(2). It is composed of large cells with an anaplastic, pleomorphic or immunoblastic cytomorphology and more than 75% of the tumour cells express the CD30 antigen(2,3). Among the other type of peripheral T-cell lymphoma, the incidence of PCALCL is 1.7% and most patients present with solitary or localised nodules, papules or plaques(3). The lesions may persist many weeks to months and reach to several centimetres, and the nodules may ulcerate over time. They can regress in 50% of patients. 20% of the patients may present with multifocal lesions. 10% of the patients will have extracutaneous dissemination(4,5). Regional lymph node involvement does not necessarily indicate the presence of systemic disease, as pathologic involvement of local nodes alone does not impact the prognosis in patients with PCALCL. Patients with involvement of regional lymph nodes and patients presenting with multifocal skin lesions have a prognosis like that of patients with only skin lesions(6).
Histologically, large CD30-positive tumour cells proliferate as diffuse cohesive sheets. There is no epidermotropism and if seen, it is particularity marked in cases of DUSP22-IRF4 rearrangement(7). The tumour cells have anaplastic features, i.e. abundant cytoplasm, round, oval or irregular shaped nuclei and prominent eosinophilic nucleoli(8).
The differential diagnosis includes Lymphomatoid papulosis (LyP), cutaneous involvement of systemic anaplastic large cell lymphoma (ALCL) and transformed mycosis fungoides(9). LyP is a chronic, recurrent, self-healing skin disease compared to PCALCL(2,9). LyP is less commonly a solitary lesion. In contrast to LyP, most cases of PCALCL present as solitary lesions; however, generalised involvement of the skin may be seen(10). Therefore, the clinical appearance and course are critical for the definite diagnosis. Lesions in LyP are usually smaller (< 1 cm) and resolve spontaneously within a few weeks or months and have a waxing and waning clinical course(11).
In this case report, the patient was a 52-year-old male with ulcerated skin tumour mass more than one cm size. This presentation was compatible with the above-mentioned studies. The CD30-positive anaplastic tumour cells were mainly in the dermis and no epidermotropism as usual cases in other studies. Unlike systemic anaplastic large cell lymphoma, PCALCL does not express EMA or ALK protein(10,12). In our case, the tumour cells were positive for CD3, CD30, MUM1 and Ki67 but negative for EMA, ALK and other markers. This was good accordance with the features of PCALCL. Patients with PCALCL should not have clinical history or evidence of mycosis fungoides. A diagnosis of mycosis fungoides with large cell transformation is likely whether tumour cells are CD30-positive or CD30-negative(13). The patient in our case report had neither clinical evidence nor clinical history of mycosis fungoides. He noticed that the solitary small tumour was developed, gradually increase in size up to 2.0 x 2.0 x 0.8 cm within three months. It also showed redness and became ulceration. Regional lymph node enlargement in left bicep muscle was noticed in three month of tumour development. The clinical course, histology and immunohistochemistry confirmed that this was involved by primary cutaneous anaplastic large cell lymphoma. Regional lymph node enlargement as an extracutaneous dissemination occurs in about 10% of patients(2,14). In our case, the patient developed the regional lymph node enlargement during three month of clinical course and its histology showed the same tumour cells of skin nodule. These cells strongly expressed CD3 and CD30.
PCALCL is an indolent disease and localised lesion can be treated with surgical excision and radiation(15). The multiple recurrence of lesions may warrant systemic therapy(5,15). Patients with widespread lesions may require multi-agent chemotherapy like CHOP(15). In our case, the patient had cutaneous lesion and regional lymph node enlargement, he got surgical excision and CHOP treatment. After 4th cycle of treatment, his lesion was cured.
Conclusion
The real incidence of PCALCL in Myanmar is unknown because of lack of reported cases. This is the first report in Myanmar and highlights the importance of early diagnosis and proper management for the disease. The definite diagnosis was made by careful histopathological examination with immunohistochemistry. Most of the cases are skin limited but it needs close monitoring due to potential risk of extracutaneous spread.
Ethical Statement
The authors have no ethical conflicts to disclose. Informed consent was obtained from the patient.
Author Contributions
Win Myat Oo performed the concept designing, laboratory diagnosis by histologic morphology, immunohistochemical study and confirmation of the case diagnosis. Sein Win performed the case investigation, case review and clinical management. Nyein Chan Aung performed the literature search and manuscript preparation. The case was conducted in Department of Clinical Haematology, Yangon General Hospital.
References
- Cesar GS, Joe Ensor, Akshjot Puri, Jasleen K Randhawa, Shilpan S Shah, Sai Ravi Kiran Pingali. Primary cutaneous anaplastic large cell lymphoma: Surveillance, Epidemiology and End results database Analysis from 2005-2016: Blood 2019 Nov 13; 134 (Supplement 1):4022.
- Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J. WHO Classification of Tumours of Hematopoietic and Lymphoid Tissues. Revised 4th ed. Lyon, France: IARC Press, 2017; pp. 395–396.
- de Oliveira LSR, Nobrega MP, Monteiro MG, et al. Primary cutaneous anaplastic large cell lymphoma-case report. An Bras Dermatol 2013;88 (6 suppl 1):132–5.
- Nerissa Moodley, Patiswa Nombona, Anisa Mosam. Primary cutaneous anaplastic large cell lymphoma-case report. Dermatopathology 2019; 6:163–169.
- Brown RA, Fernandez-Pol S, Kim J. Primary cutaneous anaplastic large cell lymphoma. J Cutan Pathol. 2017 Jun;44(6):570–7.
- Bekkenk MW, Geelen FA, van Voorst Vader PC, et al. Primary and secondary cutaneous CD30(+) lymphoproliferative disorders: a report from the Dutch Cutaneous Lymphoma Group on the long-term follow-up data of 219 patients and guidelines for diagnosis and treatment. Blood. 2000 Jun; 95(12):3653–61.
- Onaindia A, Montes-Moreno S,Rodriguez-Pinilla SM, et al. Primary cutaneous anaplastic large cell lymphomas with 6p25.3 rearrangement exhibit particular histological features. Histopathology. 2015; 66:846-55. PMID:25131361.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood.2005; 105:3768-85. PMID:15692063.
- Bolognia JL, Schaffer JV, Cerroni L, editors. Dermatology. Volume 2. 4th ed, revised. New York: Elsevier; 2018. pp. 2127–47.
- DeCoteau J, Butmarc J, Kinney M, Kadin M. The t(2;5) chromosomal translocation is not a common feature of primary cutaneous CD30+ lymphoproliferative disorders: comparison with anaplastic large-cell lymphoma of nodal origin. Blood 1996;87: 3437-41.
- Kempf W, Kerl K, Mitteldorf C. Cutaneous CD30-positive T-cell lymphoproliferative disorders-clinical and histopathologic features, differential diagnosis, and treatment. Semin Cutan Med Surg. 2018 Mar;37(1):24–9.
- de Bruin PC, Beljaards RC, van Heerde P, et al. Differences in clinical behaviour and immunophenotype between primary cutaneous and primary nodal anaplastic large cell lymphoma of T-cell or null cell phenotype. Histopathology. 1993; 23:127-35. PMID:8406384.
- Benner MF, Jansen PM, Vermeer MH, et al. Prognostic factors in transformed mycosis fungoides: a retrospective analysis of 100 cases. Blood. 2012; 119:1643-9. PMID:22160616.
- Dawn G, Morrison A, Morton R, Bilsland D, Jackson R. Co-existent primary cutaneous anaplastic large cell lymphoma and lymphomatoid papulosis.Clin Exp Dermatol. 2003 Nov;28(6):620–4.
- Liu HL, Hoppe RT, Sabine Kohler, et al., CD30+ cutaneous lymphoproliferative disorders: the Stanford experience in lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma. J Am Acad Dermatol, 2003. 49(6): p. 1049-58.