

Mantle cell lymphoma presenting as multiple lymphomatous polyposis of colon: A case report.
Paisarn Boonsakan, M.D., Suchin Worawichawong, M.D.
Department of Pathology, Faculty of Medicine Ramathibodi Hospital, Mahidol University, Bangkok, Thailand
Correspondence: Paisarn Boonsakan, M.D.
Department of Pathology, Faculty of Medicine Ramathibodi Hospital, Mahidol University, 270 Rama VI Road,
Ratchatewi, Phayathai, Bangkok, 10400, Thailand
Tel: +662-201-1432 E-mail: pboonsakan@gmail.com
Received 24 February 2014; Accepted 2 September 2014
ABSTRACT
Mantle cell lymphoma is a clinically aggressive lymphoma with a poor prognosis. Secondary involvement of gastrointestinal tract by this neoplasm is not uncommon but the primary involvement with distinguished multiple lymphomatous polyposis is unusual. We report a case of mantle cell lymphoma presenting as multiple lymphomatous polyposis of large intestine in a 57-year-old man, who presented with constipation and abdominal pain. A subsequent study showed regional lymph node and bone marrow involvement. The patient got six courses of systemic CHOP chemotherapy plus rituximab and had complete remission. Two years later, he was still alive without recurrence of the disease.
Keywords: mantle cell lymphoma, multiple lymphomatous polyposis, gastrointestinal tract lymphoma, colon
INTRODUCTION
Mantle cell lymphoma (MCL) is a distinct subtype of B-cell neoplasm, comprising approximately 3-10% of all non-Hodgkin lymphomas (NHL).1 Primary gastrointestinal (GI) tract involvement by this neoplasm is rare and ranges from 4 to 9% of all reported GI B-cell lymphomas.2, 3 Multiple lymphomatous polyposis (MLP) is the most frequent endoscopic finding in MCL. This lesion is rare and characterized by multiple polypoid lesions, involving long segments of the GI tract, and accounting for 2% of all primary GI tract lymphoma.4 We hereby report an unusual case of MCL presenting as MLP of colon, which, based on our best knowledge, is the first reported case in Thailand.
CASE REPORT
A 57-year-old Thai man without an underlying disease visited our hospital with constipation, crampy abdominal pain, and minimal mucous bloody stool for 2 days. Physical examination revealed abdominal distention and hyperactive bowel sound without evidence of peritonitis or organomegaly. Digital rectal examination demonstrated mass at the tip of finger with contact bleeding. Plain abdominal film disclosed small and large bowel dilatation with fluid level, absence of air in the rectum and absence of free air under the dome of diaphragm. Computed tomographic scan of the whole abdomen showed no evidence of bowel obstruction but thickening of intestinal wall at terminal ileum, caecum, ascending colon, proximal transverse colon, and rectosigmoid colon, including regional lymphadenopathy. A pedunculated polypoid mass at rectosigmoid region, measuring 1.9 cm in maximal dimension, was also evident. Other intraabdominal organs were not remarkable. Lymphoma or infectious process was impressed, radiologically. Laboratory studies showed no significant findings. No atypical lymphocytes were detected in peripheral blood. Gastroscopy and gastric biopsy specimen revealed no specific lesion. The colonoscopy demonstrated multiple sessile polyps, ranging from 0.5 to 1.9 cm in maximal dimension and 1 cm in average size, along the entire length of large intestine. Biopsy of a polyp found hyperplastic change (The authors could not retrieve the slide and paraffin block from the laboratory for review); therefore, familial adenomatous poly-posis/attenuated familial adenomatous polyposis was favored, clinically. Subtotal colectomy specimen was performed and gross examination of the specimen revealed multiple sessile polyps of various sizes along the colonic mucosal wall (Fig. 1). Microscopic examination demonstrated vaguely nodular and diffuse infiltration of the intestinal wall by monomorphic centrocyte-like cells (Fig. 2A and Fig. 2C). Mantle zone growth pattern was also seen (Fig. 2B). No lymphoepithelial lesion was detected, however. These neoplastic lymphoid cells showed positive immunoreactivity for CD20 (Fig. 2D), CD5 (Fig. 2E), cyclin D1 (Fig. 2F), and BCL2 and negative immunoreactivity for CD3, CD23, CD43, CD10, and BCL6. Findings supported a diagnosis of mantle cell lymphoma presenting as multiple lymphomatous polyposis. The regional lymph nodes also involved by the tumor. The subsequent workup revealed no evidence of peripheral or intrathoracic lymphadenopathy. Bone marrow biopsy demonstrated lymphomatous involvement. He was treated with CHOP chemotherapy plus rituximab (R-CHOP) and had a complete remission after six cycles of the regimen. The patient was still alive without disease progression 2 years later.
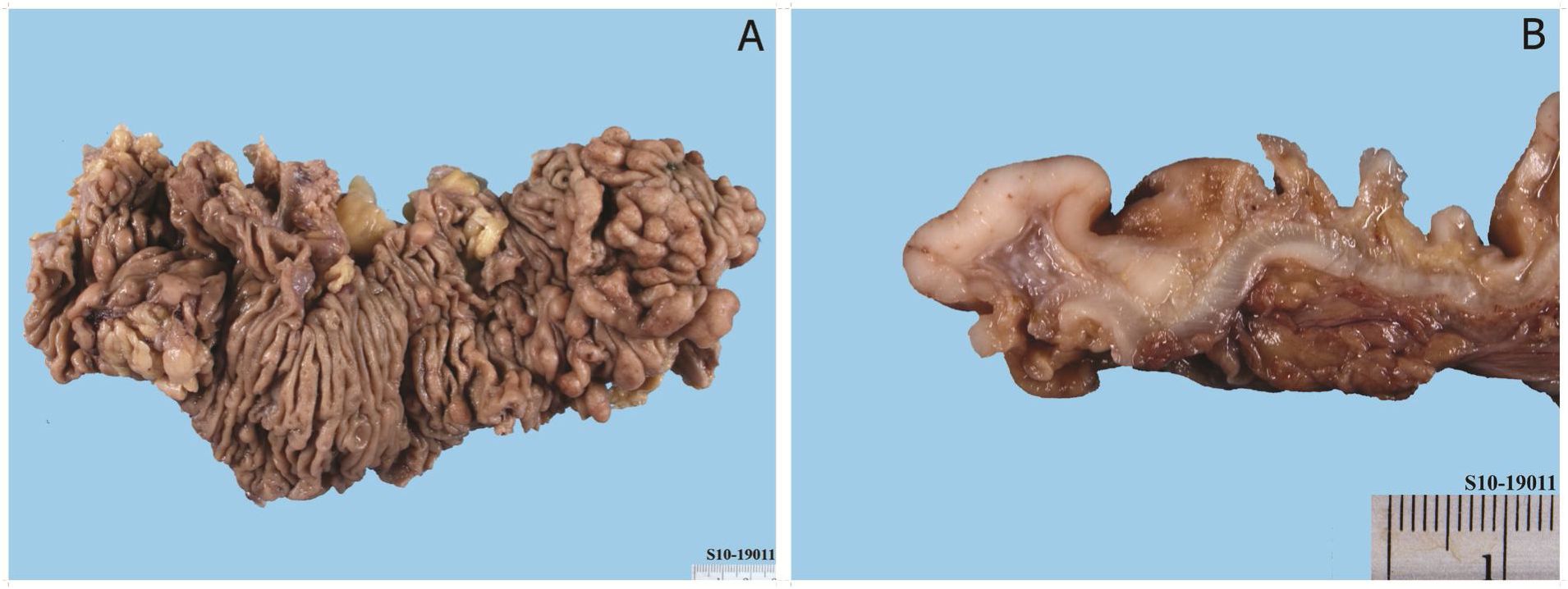
Figure 1 Mantle cell lymphoma involving the large intestine (multiple lymphomatous polyposis), gross photograph. A multiple small and large polypoid mucosal lesions. B Cut surface of the lymphoma-tous polyp showing characteristic fish-flesh appearance.
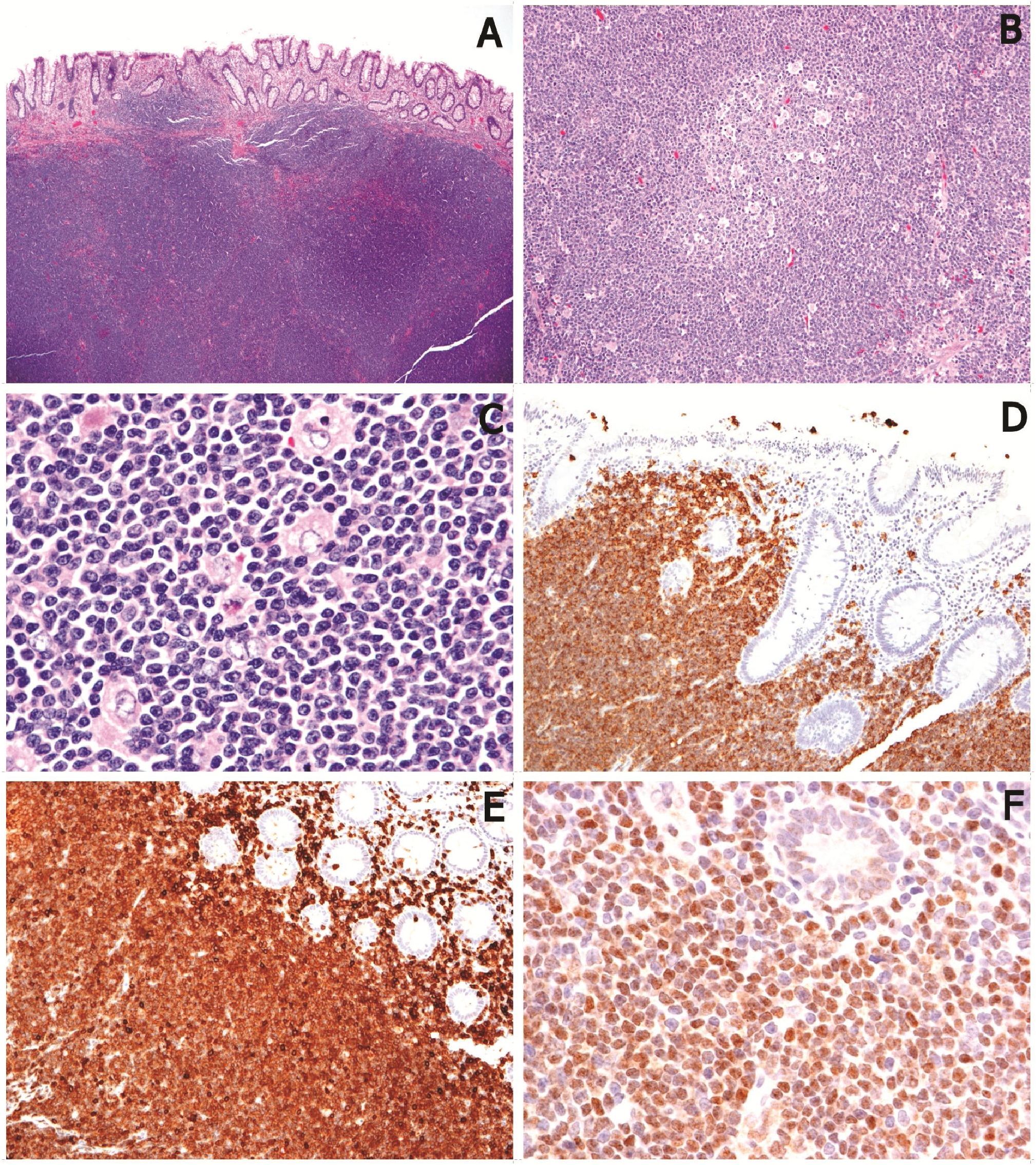
Figure 2 Histopathologic features of the lymphoma. A Vaguely nodular and diffuse intestinal mural infiltration by the lymphoma cells (H&E, original magnification X20). B Mantle zone growth pattern (H&E, original magnification XI00). C Typical neoplastic cells showing monomorphic centrocyte-like morphology admixed with scattered single epithelioid histiocytes (H&E, original magnification X400). Neoplastic cells showing positive immunoreactivity for CD20 (D),CD5 (E),and cyclin D1 (F) (original magnifications, XI00 for D, E and X400 for F)
DISCUSSION
Multiple lymphomatous polyposis (MLP), which is characterized by the presence of multiple polypoid lesions containing lymphoma cells along the GI tract, was first described by Comes in 1961.6 Mantle cell lymphoma (MCL) in a form of MLP has been reported with a frequency up to 9% of all GI B-cell lymphomas.2, 3 6-12 The disease usually occurs in male population (65%) with a mean age of 63 years.10, 11 Based on the largest study series by Ruskone-Fourmestraux A et al., the main presenting symptoms are abdominal pain, diarrhea, and hematochezia. Loss of body weight and fatigue are commonly found but gut obstruction by the tumor leading to surgery is rare. Small intestine and colon are the most common site of involvement (90% of cases), followed by rectum (60% of cases) and stomach (50% of cases). Regional lymph node involvement is not uncommon.11 Other possible affected extra-digestive tract sites include bone marrow, peripheral lymph nodes, Waldeyer’s ring, and liver.12 The endoscopic features of MLP reveal small nodular or polypoid formation ranging from 2 millimeters to severe centimeters in diameter, along the GI tract segment, with or without normal intervening mucosa.2,3
The histologic features of MCL in the GI tract include nodular, diffuse, or rarely mantle zone patterns.2,3 The neoplastic cells are rather monomor-phic and composed of mainly small to medium-sized lymphoid cells with slightly irregular nuclei (centrocytes-like cells) and scant cytoplasm. Scattering single epithelioid histiocytes are occasionally seen. 1-3 An immunohistochemical study is necessary for confirming the diagnosis. The lymphoma cells are usually positive for pan-B-cell antigens (such as CD20, CD79a), CD5, and negative for CD23, CD10, and BCL6. Almost all cases show nuclear expression of cyclin D1 protein, which is the most characteristic diagnostic criteria, and indicating an indirect evidence of translocation between IgH chain gene and the cyclin D1 (CCND1) gene resulting in overexpression of the cell cycle regulating cyclin D1.1, 2
MLP usually has advance stage of the disease at time of diagnosis.3 Primary GI tract MCL has a poor prognosis. The response rate is about 94% with the standard immunochemotherapy R-CHOP; however, the complete response rate is about 31% with relapse within 2 years.13 The macroscopic and sometimes microscopic lesions usually regressed as a response to intensive chemotherapy regimens but remission periods are short and the median survival is about 3-4 years.3 Myeloablative therapy with stem cell transplantation can significantly improve outcome in patients younger than 65 years old with good physical condition; nevertheless, at present, the current treatment protocols cannot definitively cure the patients with either peripheral or GI tract MCL.3, 14, 15 Further studies are essential for better treatment outcome.
In conclusion, we describe a rare case of primary intestinal MCL in form of MLP, predominantly involving the colon. The disease has poor prognosis and needs aggressive treatment. Although this condition is rare, it is emphasized that this entity should be included in the differential diagnosis of multiple polyposis.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGEMENT
We thank the laboratory workers in our department for skillful technical support.
REFERENCES
1. Swerdlow SH, Campo E, Seto M, Muller-Her-melink HK. Mantle cell lymphoma. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al, editors. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon: IARC; 2008. p. 229-32
2. Ruskone-Fourmestraux A, Delmer A, Lavergne A, Molina T, Brousse N, Audouin J, Rambaud JC. Multiple lymphomatous polyposis of the gastrointestinal tract: prospective clinicopatho-logic study of 31 cases. Groupe D’etude des Lymphomes Digestifs. Gastroenterology. 1997 Jan;112:7-16.
3. Ruskone-Fourmestraux A, Audouin J. Primary gastrointestinal tract mantle cell lymphoma as multiple lymphomatous polyposis. Best Pract Res Clin Gastroenterol. 2010;24:35-42.
4. Meral M, Demirpen^e M, Gonen C, Akarsu M, Kayahan H, Demirkan F, et al. Diffuse gastrointestinal involvement of mantle cell lymphoma. Turk J Gastroenterol. 2008;19:117-20.
5. Cornes JS. Multiple lymphomatous polyposis of the gastrointestinal tract. Cancer 1961; 14: 24957
6. Salar A, Juanpere N, Bellosillo B, Domingo-Do-menech E, Espinet B, Seoane A, et al. Gastrointestinal involvement in mantle cell lymphoma: a prospective clinic, endoscopic, and pathologic study. Am J Surg Pathol. 2006;30:1274-80.
7. Chen CY, Wu CC, Hsiao CW, Lee TY, Jao SW. Colonic manifestation of mantle cell lymphoma with multiple lymphomatous polyposis. Int J Colorectal Dis. 2009;24:729-30.
8. Michopoulos S, Petraki K, Matsouka C, Kastri-tis E, Chrysanthopoulou H, Dimopoulos MA. Mantle-cell lymphoma (multiple lymphomatous polyposis) of the entire GI tract. J Clin Oncol. 2008;26:1555-7.
9. Moussaide G, Kazemi A, Mitre R, Mitre MC. Mantle cell lymphoma: a rare cause of a solitary duodenal mass. BMJ Case Rep. 2014;23:1-3.
10. Remes-Troche JM, De-Anda J, Ochoa V, Barreto-Zuniga R, Arista-Nasr J, Valdovinos MA. A rare case of multiple lymphomatous polyposis with widespread involvement of the gastrointestinal tract. Arch Pathol Lab Med. 2003;127:1028-30.
11. Ruskone-Fourmestraux A, Delmer A, Lavergne A, Molina T, Brousse N, Audouin J, et al. Multiple lymphomatous polyposis of the gastrointestinal tract: prospective clinicopathologic study of 31 cases. Groupe D’etude des Lymphomes Digestifs. Gastroenterology. 1997;112:7-16.
12. Chung Kim Yuen C, Tomowiak C, Yacoub M, Barrioz T, Barrioz C, Tougeron D. A rare case of mantle cell lymphoma as lymphomatous polyposis with widespread involvement of the digestive tract. Clin Res Hepatol Gastroenterol. 2011;35:74-8.
13. Lenz G, Dreyling M, Hoster E, Wormann B, Duhrsen U, Metzner B, et al. Immunochemo-therapy with rituximab and cyclophosphamide, doxorubicin, vincristine, and prednisone significantly improves response and time to treatment failure, but not long-term outcome in patients with previously untreated mantle cell lymphoma: results of a prospective randomized trial of the German Low Grade Lymphoma Study Group (GLSG). J Clin Oncol. 2005;23:1984-92.
14. Murali S, Winton E, Waller EK, Heffiier LT, Lo-nial S, Flowers C, et al. Long-term progression-free survival after early autologous transplantation for mantle-cell lymphoma. Bone Marrow Transplant. 2008;42:529-34.
15. Romaguera JE, Fayad L, Rodriguez MA, Bro-glio KR, Hagemeister FB, Pro B, et al. High rate of durable remissions after treatment of newly diagnosed aggressive mantle-cell lymphoma with rituximab plus hyper-CVAD alternating with rituximab plus high-dose methotrexate and cytarabine. J Clin Oncol. 2005 Oct 1;23(28):7013-23.