

Congenital pulmonary airway malformation of lung: a report of three cases
Smita Singh, Kiran Agarwal and Neha Suman*
Department of Pathology, Lady Hardinge Medical College, New Delhi, India
Correspondence to:
Dr Neha Suman,
Department of Pathology, Lady Hardinge Medical College,
Shaheed Bhagat Singh Marg, New Delhi 110001, India.
Telephone: +91 (0) 11 2336 3728 Email: drnehasuman@gmail.com
Conflict of interest: The authors declare that they have no conflicts of interest with the contents of this article.
|
Submitted: |
12 February 2021 |
|
Accepted: |
20 February 2021 |
|
Published: |
26 March 2021 |
Abstract
Congenital pulmonary airway malformation (CPAM), also known as congenital cystic adenomatoid malformation is a rare congenital anomaly of lung involving the foetal tracheobronchial tree. It is a rare cause of respiratory distress in neonates. The reported incidence of this entity is 1 in 11,000 to 1 in 35,000. Here we present three such cases of CPAM because of its rarity and also to emphasise on the cystic diseases of lung. We also briefly summarise the CPAM subtypes and differential diagnosis of cystic lung diseases of childhood. Correct diagnosis is important for better management and hence better outcome in patients.
Keywords: congenital pulmonary airway malformation; neonate; respiratory distress
Introduction
Congenital pulmonary airway malformation (CPAM), also known as congenital cystic adenomatoid malformation is a rare congenital anomaly of lung, first described by Chin and Tang in 1949(1). The reported incidence of this entity is 1 in 11,000 to 1 in 35,000. The patients commonly present with respiratory distress, recurrent chest infection and haemoptysis(2). The exact aetiology is not known but there is evidence suggestive of arrested lung development between 35 and 55 days of gestation(3). Here we report 3 interesting cases of CPAM – one case of CPAM Type 1 and two cases of CPAM Type 2 (Stocker's classification), along with a brief summary of CPAM subtypes according to Stocker's classification and differential diagnosis of congenital cystic lung diseases.
Case 1
A one-year-old male baby presented with the chief complaint of respiratory distress in the paediatric emergency. On examination, tachypnoea was present. He had history of recurrent chest infections since birth. Computed tomography (CT) scan of the thorax showed multicystic lesion involving the upper lobe of the right lung. He underwent lobectomy. Grossly, the resected lobe measured 9 x 6 x 2 cm. External surface was light brown, congested and pleura was thinned out. Cut section was spongy in texture and showed multiple cysts, largest measuring 5.5 cm in greatest dimension. Normal lung parenchyma was identified. Microscopy revealed cysts resembling abnormal bronchiole like structures. These cysts were lined by ciliated cuboidal epithelium resting on thin fibromuscular wall (Figure 1). Based on gross feature and histology, the final diagnosis of CPAM of the lung (Stocker Type 2) was made. Also, there were features of emphysema and interstitial pneumonitis in the adjacent lung. The post-operative period of the patient was uneventful.
Case 2
A five-month-old male baby presented with respiratory distress. He had significant history of recurrent chest infections since birth. The clinical history revealed that the antenatal ultrasonography of the mother was suggestive of cystic lesion in the left upper lobe of lung of the foetus. The patient underwent CT scan of the chest which revealed cystic lesions involving the upper lobe of the left lung. Left-sided lobectomy was done. The post-operative period was uneventful. Grossly, the lobectomy specimen measured 8.5 x 6.5 x 2 cm. There was presence of single large cyst measuring 7 x 6 cm with wall thickness varying from 0.2 to 0.3 cm. Also, few smaller cysts varying in size from 0.2 to 0.5 cm were noted. Normal lung tissue was identified at the periphery. On microscopic examination, these cysts were lined by pseudostratified ciliated columnar epithelium (Figure 2). The diagnosis of CPAM of the lung (Stocker Type 1) was confirmed.
Case 3
A one-day-old male infant presented with respiratory distress since birth. On examination, tachypnoea and diminished breath sounds in the left lower lung field were present. There was history of prolonged labour and the antenatal ultrasonography of the mother was suggestive of cystic lesion in left lung of the foetus. He underwent CT scan of the chest which revealed multicystic lesions involving the lower lobe of left lung. Left-sided lobectomy was done. Gross appearance of the resected lobe showed smooth outer surface covered with pleura. It measured 7 × 6.5 × 1.4 cm. Cut surface showed 3 cysts, largest measuring 5 cm in maximum dimension. Microscopically, variable-sized intercommunicating cysts resembling bronchioles were seen. These cysts were lined by ciliated cuboidal epithelium (Figure 3). Focal areas showed features of emphysema and meconium in adjacent alveoli. The diagnosis of CPAM of the lung (Stocker Type 2) was made. The post-operative period of the patient was uneventful.
Discussion
CPAM is a rare malformation of lung, presenting usually in children within first 2 years of life. Antenatal ultrasound plays a very important role in diagnosis of this congenital anomaly. Approximately one-third of the antenatally diagnosed lesions resolve before birth(4). The exact aetiology of CPAM is not known, however, few reports describing the probable aetiopathogenesis have been documented. Embryogenic insults occurring between 35 and 55 days of gestation leads to abnormal development of terminal bronchiolar structures of the lung. Many genes are implicated in the development of lung-thyroid transcription factor gene (Nkx2), sex-determining region Y-box 2 gene (Sox2), Hox gene (Hoxb-5), Ying Yang 1 gene (Yy1), fatty acid-binding protein-7 gene (FABP-7), acyl-CoA synthetase 5 gene (ACSL5), platelet-derived growth factor B gene (PDGF-B), sonic hedgehog gene (SHH), bone morphogenetic protein 4 gene (BMP4), sprouty 2 gene (SPRY2), Wnt signalling pathways, transforming growth factor beta gene (TGF-ß) and fibroblast growth factors 7, 9 and 10 genes (FGF7, 9 and 10). All of these genes have a role in cell proliferation or apoptosis, leading to the various types of malformations classified under the CPAM classification. These genes are involved in cell proliferation or apoptosis during various stages of embryogenesis. Any defect in these genes result in imbalance in cell proliferation and apoptosis leading to malformation of lung(3,5,6). Bronchography studies in these cases have revealed that bronchial atresia is the primary defect leading to the development of CPAM. The type of malformation is based on abnormal lung growth beyond the atretic segment.
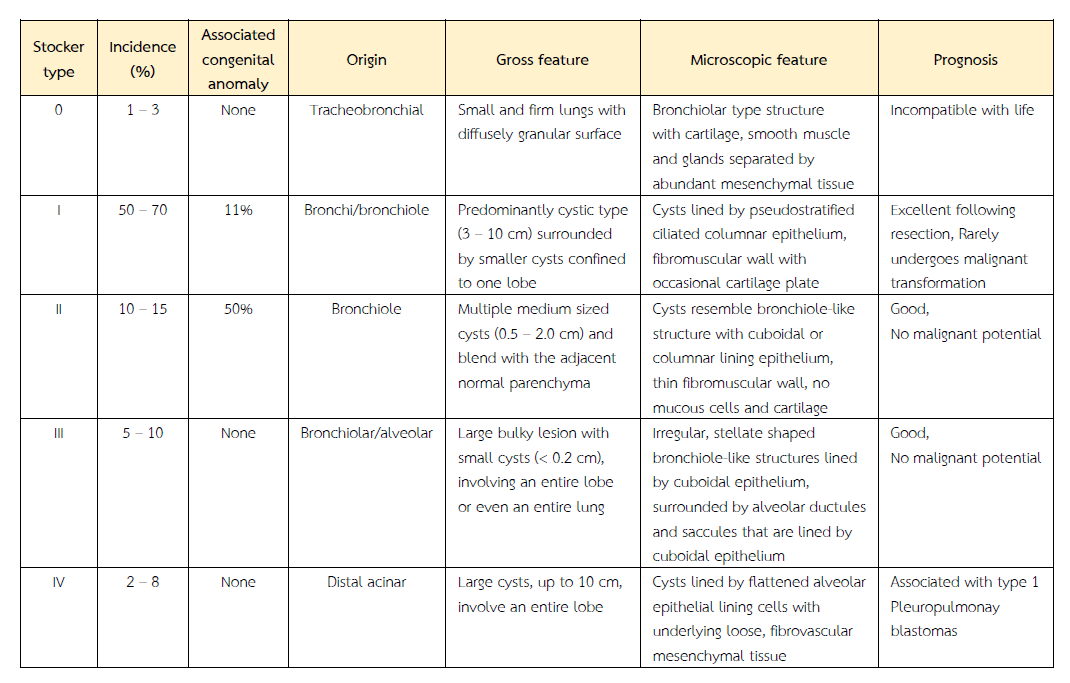
The patients usually present with respiratory distress in approximately 80% of the cases. It is secondary to pulmonary hypoplasia or compression. Recurrent chest infections, pneumothorax and rare instances of haemopneumothorax are also seen. It is usually restricted to one lobe (as observed in our cases), but can involve whole of one lung, or both the lungs. The most acceptable classification used for diagnosis and treatment was published by Stocker (Table)(7). In our report, one case was of CPAM type 1 and two cases belonged to Type 2. Antenatal ultrasonography had revealed cystic lung lesion in two cases. All the three cases were males and they presented with respiratory distress.
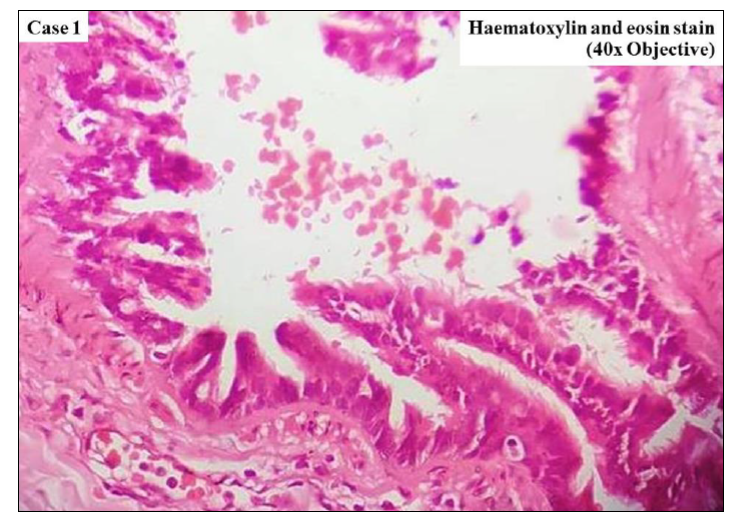
Figure 1 Cysts lined by ciliated cuboidal epithelium, resembling bronchiole [Haematoxylin and eosin (H&E), ×400].
Imaging studies – chest x-ray and CT are helpful in diagnosing the cystic lesions of lung but the confirmatory diagnosis of CPAM is made only by histopathological examination. Bronchopulmonary sequestration (BPS) is a very important differential diagnosis and its exclusion especially in cases of Type 3 CPAM is very crucial. Both Type 3 CPAM and BPS are solid-appearing echogenic masses with well-defined borders. However, BPS has no connection to the tracheobronchial tree. It receives its blood supply from the systemic circulation via an anomalous systemic artery rather than the pulmonary circulation, as in CPAM(8). Other differential diagnoses to consider are congenital diaphragmatic hernia (CDH), bronchogenic cyst, congenital lobar over-inflation (CLO), primary ciliary dyskinesia (PCD), cystic fibrosis (CF) and Williams-Campbell syndrome (WCS)(8-11). In CDH, the herniated organ is intestine. Bronchogenic cysts are usually isolated lesions originating from the upper airway. There is unilocular thin walled cyst with smooth surface. Microscopically, the cyst wall is lined by ciliated pseudostratified columnar cells. Hyaline cartilage, seromucinous glands and smooth muscle are seen in the cyst wall. The absence of cartilage in CPAM excludes bronchogenic cyst(8). Congenital lobar over-inflation (CLO) also known as congenital lobar emphysema shows mild alveolar dilatation without destruction of the alveolar septa(12). In PCD, there is abnormality in the structure or function of the cilia resulting in mucus accumulation in the lungs. Bronchial dilation is present secondary to mucus accumulation(13). CF is an autosomal recessive disorder, with primary defect in the function or absence of cystic fibrosis transmembrane conductance regulator (CFTR) gene. There is thick viscous mucus leading to obstruction of exocrine glands and bronchi lumens. Features of brochiectasis, emphysema and abscess secondary to infections can be seen in CF(14). WCS is a rare congenital syndrome characterised by cystic lung lesions associated with absence of cartilage in the subsegmental bronchi leading to bronchiectasis distal to the affected bronchi. There is bilateral involvement of lungs along with associated anomalies such as congenital cardiac diseases. Microscopic examination reveals absence of cartilage in the bronchial wall(15). Other cystic lesions in lung are cystic teratoma and enteric duplication cyst. Cystic teratoma contains derivatives from all the three germ layers(11). Enteric (oesophageal) duplication cyst is lined by stratified squamous or gastrointestinal epithelium and contains smooth muscle layer. These entities may also be considered in the differential.
Early pathological diagnosis is very important in congenital cystic lung diseases to determine the prognosis. Surgical resection – lobectomy or non-anatomical segmentectomy is mostly recommended for management of CPAM. Early surgery may help in adequate growth and expansion of the remaining lung to restore the total lung volume and ensure satisfactory pulmonary function(16).
Conclusion
To summarise, CPAM is a rare congenital malformation of lung presenting with respiratory distress in most of the cases. Proper histopathological examination is crucial for excluding other differential diagnosis and rendering correct diagnosis. The classification of the lesion further helps in management and allows assessing the prognosis of the patients.
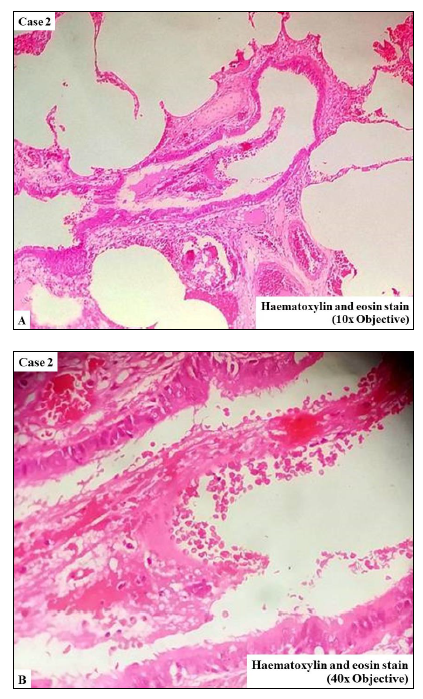
Figure 2 Cysts lined by pseudostratified ciliated columnar epithelium. A (H&E, x100); B (H&E, x400)
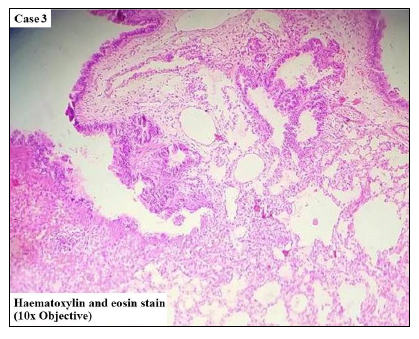
Figure 3 Cysts lined by ciliated cuboidal epithelium, resembling bronchiole (H&E, ×100).
Table Stocker's classification of congenital cystic adenomatoid malformation of lung.
References
- Ch'in KY, Tang MY. Congenital adenomatoid malformation of one lobe of a lung with general anasarca. Arch Pathol 1949; 48:221-9.
- Gornall AS, Budd JL, Draper ES, Konje JC, Kurinczuk JJ. Congenital cystic adenomatoid malformation: Accuracy of prenatal diagnosis, prevalence and outcome in a general population. Prenat Diagn 2003; 23:997-1002.
- Laberge JM, Flageole H, Pugash D, Khalife S, Blaie S, Filiatraut D, et al. Outcome of the prenatally diagnosed congenital cystic adenomatoid lung malformation: A Canadian experience. Fetal Diagn Ther 2001; 16:178-86.
- Davenport M, Warne SA, Cacciaguerra S, Patel S, Greenough A, Nicolaides K. Current outcome of antenatally diagnosed cystic lung disease. J Pediatr Surg 2004; 39:549-56.
- Moerman P, Fryns JP, Vandenberghe K, Devlieger H, Lauweryns JM. Pathogenesis of congenital cystic adenomatoid malformation of the lung. Histopathol 1992; 21: 315-21.
- David M, Lamas-Pinheiro R, Henriques-Coelho T. Prenatal and Postnatal Management of Congenital Pulmonary Airway Malformation. Neonatology. 2016; 110:101-15.
- Stocker JT. Congenital pulmonary airway malformation: A new name and an expanded classification of congenital cystic adenomatoid malformations of the lung. Histopathol 2002; 41: 424-31.
- Sfakianaki AK, Copel JA. Congenital cystic lesions of the lung: congenital cystic adenomatoid malformation and bronchopulmonary sequestration. Rev Obstet Gynecol. 2012; 5:85-93.
- Durell J, Lakhoo K. Congenital cystic lesions of the lung. Early Hum Dev. 2014; 90:935-9.
- Ursini WP, Ponce CC. Congenital pulmonary airway malformation. Autops Case Rep. 2018; 8(2):e2018022.
- Barman S, Mandal KC, Kumar R, Biswas SK, Mukhopadhyay M, Mukhopadhyay B. Congenital cystic lesions of lung in the paediatric population: A 5-year single institutional study with review of literature. Afr J Paediatr Surg 2015; 12:66-70.
- Cataneo DC, Rodrigues OR, Hasimoto EN, Schmidt AF Jr, Cataneo AJ. Congenital lobar emphysema: 30-year case series in two university hospitals. J Bras Pneumol. 2013; 39:418-26.
- Mirra V, Werner C, Santamaria F. Primary ciliary dyskinesia: an update on clinical aspects, genetics, diagnosis, and future treatment strategies. Front Pediatr. 2017; 5:135.
- Jain M, Goss CH. Update in cystic fibrosis 2013. Am J Respir Crit Care Med. 2014; 189:1181-6.
- Wayne KS, Taussig LM. Probable familial congenital bronchiectasis due to cartilage deficiency (Williams-Campbell syndrome). Am Rev Respir Dis. 1976; 114:15-22.
- Baird R, Puligandla PS, Laberge JM. Congenital lung malformations: informing best practice. Semin Pediatr Surg. 2014; 23:270-7.