

Pulmonary alveolar proteinosis coexisting with homozygous hemoglobin E
Niramol Chanplakorn1, Atcharaporn Pongtippan1, Harutai Kamalaporn2
Department of Pathology, Faculty of Medicine, Ramathibodi Hospital, Mahidol University, Bangkok, Thailand 2
Department of Pediatrics, Faculty of Medicine, Ramathibodi Hospital, Mahidol University, Bangkok, Thailand
ABSTRACT
Background: Pulmonary alveolar proteinosis (PAP) is an uncommon lung disorder of obscure and possibly diverse causes characterized by abnormal accumulation of lipoproteinaceous surfactant material in alveoli. The prevalence of acquired PAP has been estimated to be 0.37 per 100,000 populations. We present a case of PAP coexisting in homozygous hemoglobin (Hb) E.
Case presentation: A 13-year-old girl who had been diagnosed as homozygous Hb E. She presented with dyspnea and cough. Thoracotomy was performed due to abnormal mediastinal mass disclosed in CT scan. However, only slightly thymus gland enlargement was found then wedge lung biopsy was performed. Examination of the sections of lung revealed alveoli diffusely filled with eosinophilic granular material. Bron-choalveolar lavage was also performed. Cytopathologic evaluation revealed a milky appearance with microscopically amorphous granular material with positive periodic acid-Schiff stain and scant chronic inflammatory cells. Findings were consistent with PAP. The clinical condition of the patient was stable after treatment.
Keywords: pulmonary alveolar proteinosis, bronchoalveolar lavage, cytology, hemoglobin E
Correspondence:
Niramol Chanplakorn, M.D., Ph.D.
Department of Pathology, Faculty of Medicine, Ramathibodi Hospital, Mahidol University
270 Rama VI Road, Ratchatewi, Phayathai, Bangkok, Thailand 10400
Tel+662-201-1432, 662-201-1437, 662-201-1667
E-mail: drniramol_ch@yahoo.com, rancp@mahidol.ac.th
INTRODUCTION
Pulmonary alveolar proteinosis (PAP) is a rare disorder characterized by the accumulation of surfactant proteins and phospholipids within the alveolar spaces. PAP can be classified into congenital, secondary and acquired forms (1). Acquired PAP is divided into two forms on the basis of clinical features: idiopathic PAP and secondary PAP. Idiopathic acquired PAP was originally defined in 1958 (2) and it comprises more than 90 % of cases (1). Secondary PAP is related to a number of clinical situations, including pharmacologic immunosuppression, dust exposure or toxic fumes, infections, and hematologic malignancy (1-4). The prevalence of idiopathic PAP has been estimated to be 0.37 per 100,000 persons (1). The median age at the time of diagnosis is 39 years (1). The series reported in the literature suggest a male predominance (male: female ratio 3:1) that may be linked to tobacco smoking (1, 5). Several recent studies in transgenic mice and human have demonstrated that impairment of surfactant clearance by alveolar macrophages as a result of inhibition of the action of granulocyte-macrophage colony-stimulating factor (GM-CSF) by blocking autoantibidies may underlie many acquired idiopathic PAP. Secondary PAP may result from functional impairment of GM-CSF receptor on alveolar macrophages and/or an abnormal signal transduction pathway after interaction of GM-CSF and its receptor that is associated with hematologic disorders (1, 3-4).
A diagnosis of PAP is based on the examination of bronchoalveolar lavage (BAL) specimen and/or an examination of lung biopsy. Cytologic findings of BAL specimens from patients with PAP have been reported (6-7) but most reports come from western countries and reports in Asian populations are rare (4, 6-12).
Hemoglobin (Hb) E is very common in parts of Southeast Asia and expresses varied mutation. It results in a heterogenous group of disorders whose phenotype range from asymptomatic to severe. Hb E trait and homozygous Hb E are mild disorders (13-14). Although secondary PAP associated with hematologic disorders such as chronic granulocytic leukemia with progressive myelofibrosis, myeloproliferative disorders and myelodysplastic syndrome (3-4, 9, 12) has been reported, but coexistence of acquired PAP with hemoglobinopathy, in particular homozygous Hb E, has never been reported to the best of our knowledge.
In this paper, we report a case of homozygous Hb E coexisting with acquired PAP which was diagnosed by histological findings of lung biopsy and subsequently whole lung lavage for treatment. Case report
A thirteen-year-old girl, known case of Hb E (Hb baseline 13.5 g/dL), was referred to our hospital in 2009 from a local hospital in Chonburi province. She presented with dyspnea and cough for one month. The provisional diagnosis from that local hospital was severe pneumonia with a suspected of pulmonary tuberculosis. Then antituberculosis and antibiotics treatment had been given for two months but her clinical condition was getting worse. The CT scan of chest found anterior mediastinal mass with extensive increased opacification with air-bronchogram and bronchiologram in both lungs. Therefore, thoracotomy was performed and revealed slightly thymus gland enlargement, then wedge lung biopsy was concurrently performed. Examination of sections obtained from left upper lobe revealed alveoli diffusely filled with eosinophilic granular material admixed with rare cholesterol cleft, however, no evidence of inflammation or vasculitis was found.
She was referred to our hospital for further management. Physical examination on the first admission, pale conjunctiva but no clubbing of fingers was noted. Bilateral fine crepitation was audible at both chest walls. Spleen was also palpable 1 cm. below left costal margin. No other abnormality was detected. Ultrasonography of abdomen revealed splenomegaly without other focal mass. Laboratory examinations showed decreased in hemoglobin levels but other serologic study showed negative antiHIV, negative for mycoplasma antibody, and negative blood PCR study for CMV. CMV viral load was less than 600 copies per milliliter of blood plasma. Ig A, Ig G, and Ig M level were within normal range. Blood antinuclear antibody (ANA) test showed positive at 1:80 dilution with fine speckled pattern. C3 and C4 level were within normal range and negative p-antineutrophil cytoplasmic antibody (ANCA) and c-ANCA. High-resolution CT study revealed decreased air-space with consolidation patchy geographic area of ground-glass opacity.
BAL with whole lung lavage was performed for cytologic study and treatment, respectively, and it also showed negative for ANA test. BAL specimen was opaque milky in appearance and revealed a large amount of amorphous granular cyanophilic and basophilic extracellular material with few alveolar macrophages and lymphocytes (figure 1). This material showed brightly granular stained for periodic acid-schiff (PAS) reaction, with or without diastase digestion (figure 2). Cytologic findings were compatible with pulmonary alveolar proteinosis. Initial serum lactate dehydrogenase (LDH) level was 776 U/L and the level was consequentially decreased after BAL treatment to 549U/L on second week of admission, 409 U/L on third week of admission and 175 U/L before discharge from hospital. In addition, obtained sections of lung biopsy specimen were reviewed and the histologic findings revealed structurally intact alveoli diffusely filled with eosinophilic granular material (figure 3 and 4). This material was also positive for PAS stain. No cholesterol cleft was found. Neither viral-cytopathic effect nor fungal infection was found in PAS and Grocott methenamine silver (GMS) stains. The ultrastractural study by transmission electron microscopy (TEM) showed lamellar bodies (Figure 5) Findings were compatible with alveolar proteinosis. However, further investigation for surfactant protein A (SP-A) or granulocyte macrophage colony-stimulating factor (GM-CSF) analysis measurement of GM-CSF level in BAL fluid or blood could not be performed because unavailable laboratory for these tests in Thailand. According to the proper clinical setting and results of investigations, the patient was diagnosed as acquired pulmonary alveolar proteinosis.
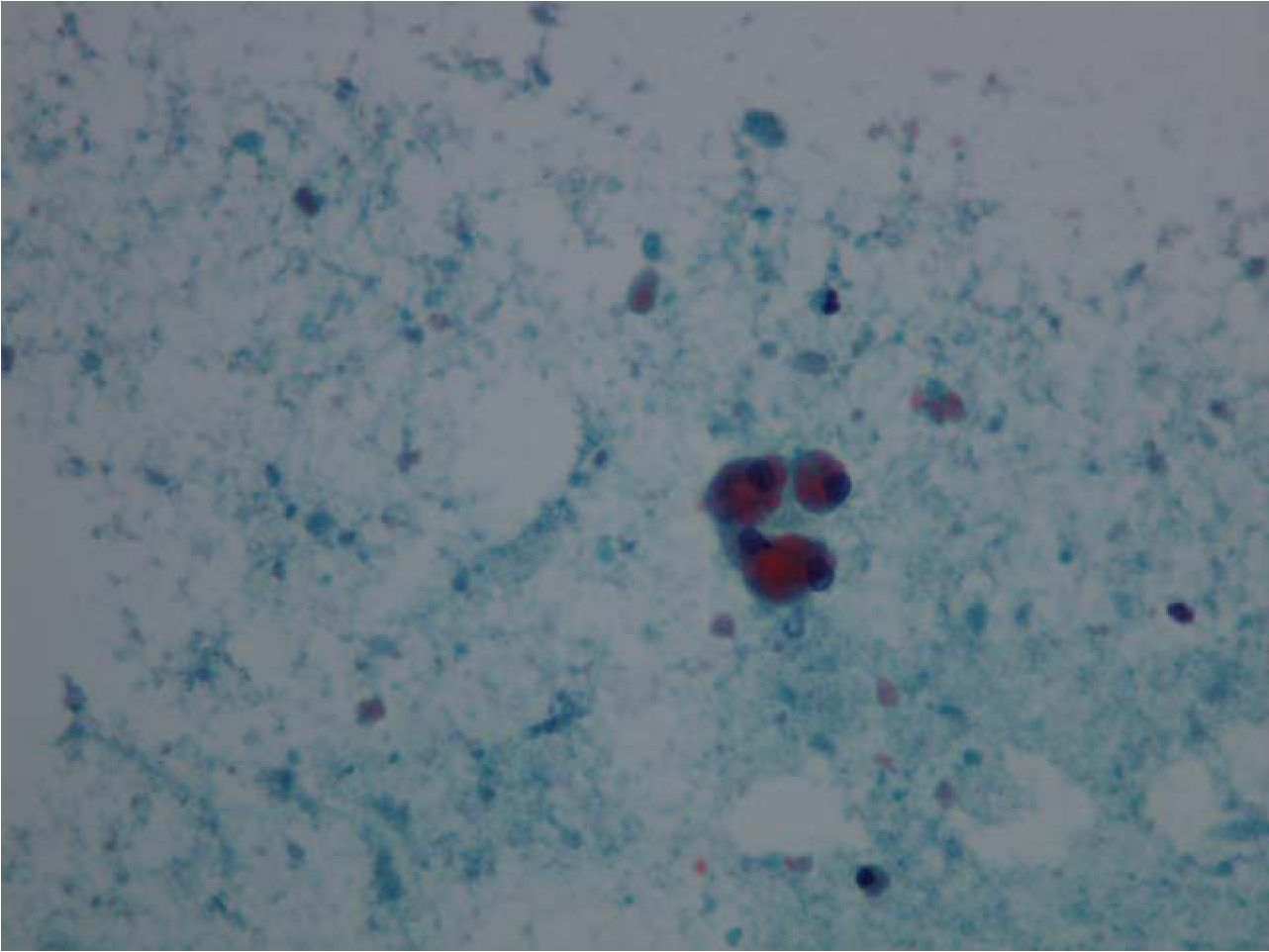
Figure 1 Papanicolaou-stained smear of bronchoalveolar lavage fluid shows mainly amorphous granular and globular extracellular material with few foamy alveolar macrophages (original magnification x400).
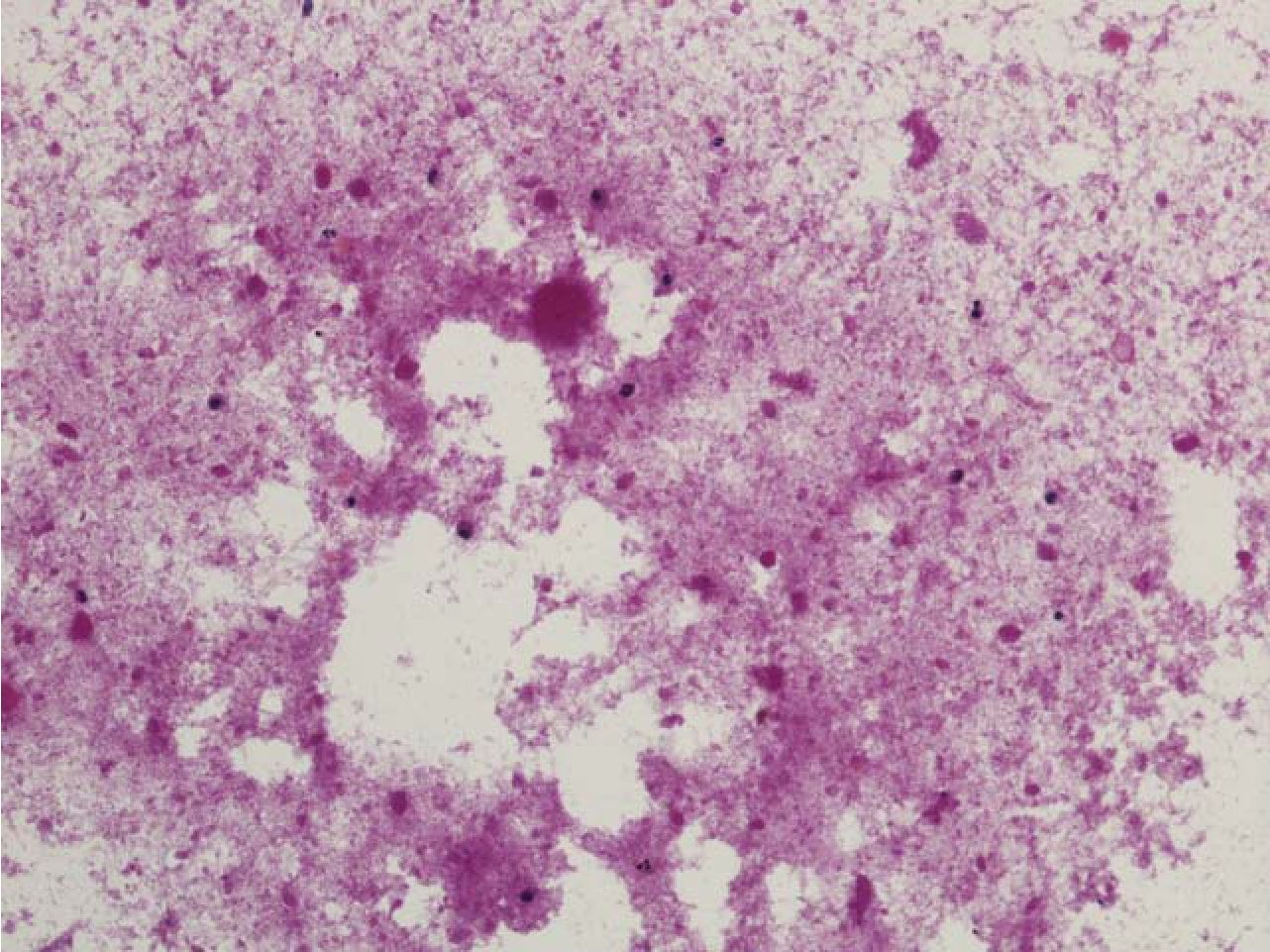
Figure 2 The globular material shows strong PAS stain (original magnification x100).
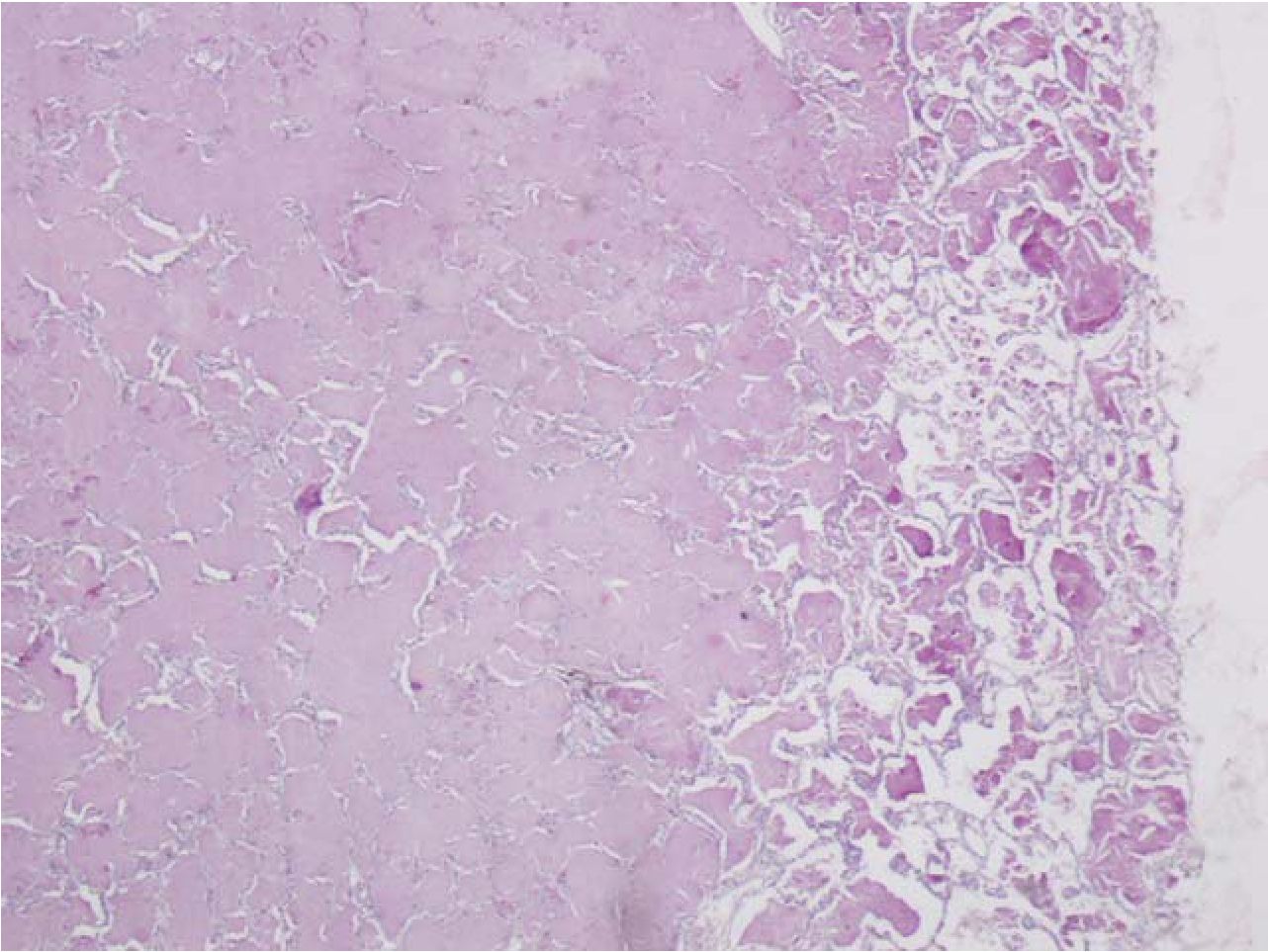
Figure 3 Section of the open lung biopsy reveals alveoli diffusely filled with abundant granular eosinophilic material with remarkable large lamellar concretions within the alveoli near pleural surface (Hematoxylin-eosin-stained, original magnification x40).
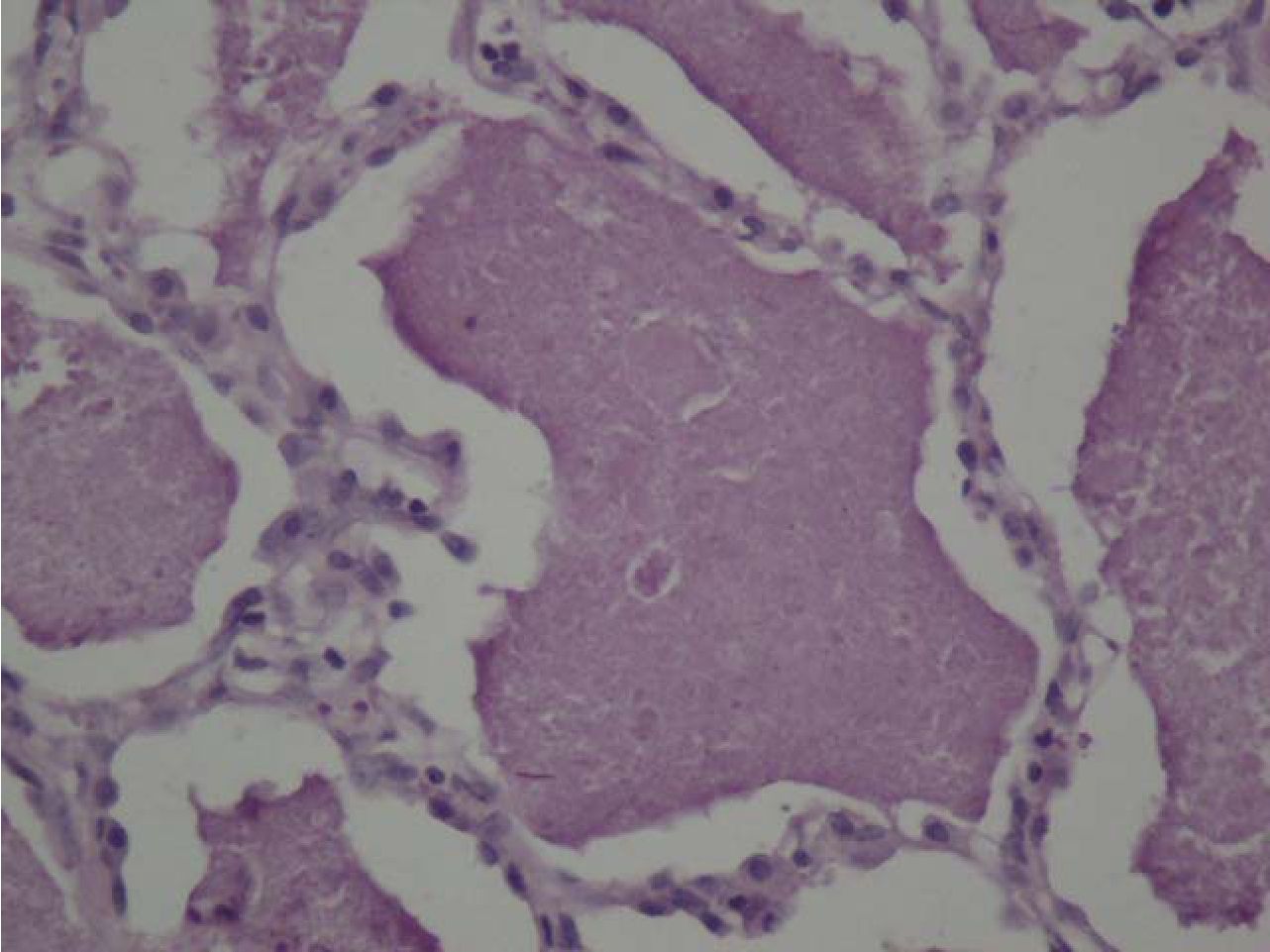
Figure 4 Section of the open lung biopsy reveals alveoli diffusely filled with abundant granular eosinophilic material with scant alveolar macrophages (original magnification x400).
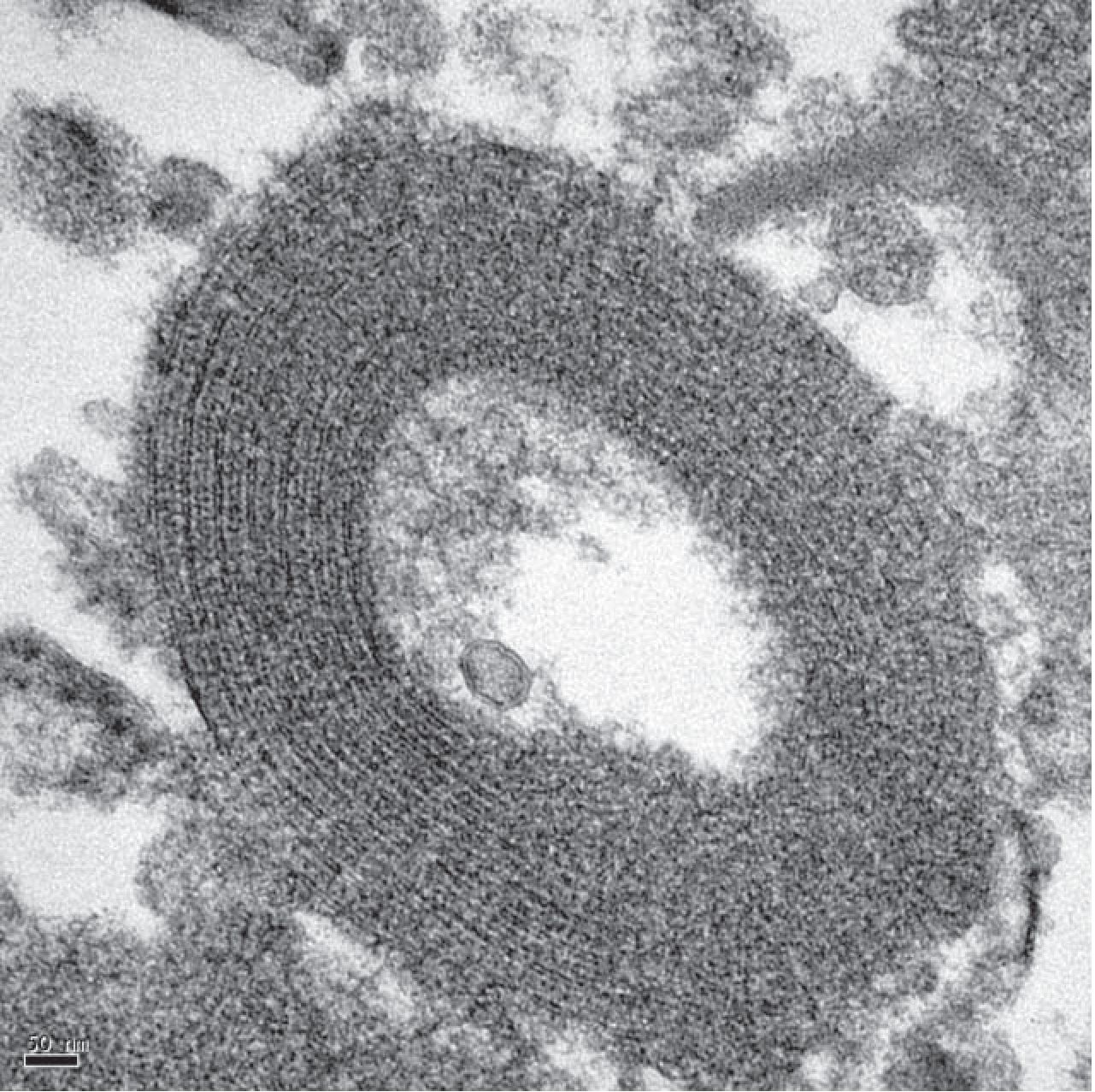
Figure 5 The ultrastructural study of BAL demonstares lamellar body which is characteristic of alveolar proteinosis. (TEM X 40,000)
DISCUSSION
PAP is an uncommon lung disease and is included in the differential diagnosis of patients all ages presenting with dyspnea. PAP was first described in 1958 by Rosen and colleagues (2) as a rare lung disorder in which the alveoli are filled with PAS-positive proteinaceous material, rich in lipid. The exact etiology is not clear and probably is multifactorial (1-8). Initially, it was thought that an inhaled irritant (e.g., silica) or infectious agent induced an excessive production of the natural material lining the alveoli caused PAP (10). However, the inability to find such agents in the lung-biopsy specimens of most PAP patients failed to support this idea (1). Recent observations in animal experiments and human suggest that the acquired PAP caused by abnormal surfactant homeostasis due to presence of the neutralizing anti-GM-CSF autoantibodies (1, 5). These autoantibodies against GM-CSF cause defects in the functioning of alveolar macrophages, including impairment of the catabolism of surfactant lipids and proteins and disruption of surfactant homeostasis (1, 5).
Although in the past a diagnosis was made by open lung biopsy, but in recent year BAL cytology and/or transbronchial biopsy have been useful for the diagnosis. Electron microscopy (EM) demonstrating lamellar bodies can provide ultrastructure confirmation (7, 11). However, this expensive and time-consuming procedure is not available to many hospitals and is unnecessary in most cases. Recently, BAL with lower cost and convenience has become the standard for both the diagnosis and treatment for PAP (6, 7, 11). On cytologic examination of BAL specimen based on Papanicolaou’s stain reveals amorphous, globular, cyanophilic or basophilic PAS-positive diastase resistant material, generally, with low numbers of inflammatory cells and macrophages. This globular material of BAL specimen can be of significant value in making a diagnosis of PAP, especially when the number of globules is more than 18 (6).
In differential diagnosis, some of the conditions to be considered include pulmonary edema, pneumocystis pneumonia, amyloidosis, amiodar-one toxicity, and other pulmonary infections such as nocardiosis, histoplasmosis, cryptococcosis, aspergillosis, mycobacterial diseases, candidiasis, and multiple viral infections (6-7, 11). Pulmonary edema lacks the globular material and cellular debris seen in PAP. Pneumocystis pneumonia can be distinguished from PAP by a more dramatic clinical presentation, exudates that appear finely vacuolated or foamy, and significantly more inflammatory background. With carefully search, the exudates are composed of one or two tiny dot-like trophozoites, measuring 0.5 to 1 pm in diameter, which could be confirmed by GMS stain. Amyloidosis has also been rarely reported in BAL specimen and amyloid usually has a “harder”, more discrete appearance than fluffy amorphous material of PAP (7). The Congo red stain produces a characteristic reddish color with yellowish-green birefringence for amyloid. Amiodarone toxicity is other rare potential causes of false-positive clinical and cytologic diagnoses of PAP (7, 11). The clinical manifestations of pulmonary toxicity include progressive dyspnea and cough. BAL specimen reveals many large mono and multinucleated macrophages with finely foamy cytoplasm. Osmophilic lamellar inclusion bodies have also been observed in lysosomes of alveolar macrophages by EM, corresponding to foamy cytoplasm in macrophages by light microscopy that are similar to the ultrastructural appearance of PAP (5).
In conclusion, we report a case of homozygous Hb E coexisting with acquired PAP that is rarely found in Thailand. The coexisting between PAP and hemoglobinopathy in this case is still unexplainable and await for further investigation.
ACKNOWLEDGEMENT
We thank Dr. Woranart Ratanakorn, Dr. Ratana Tantranon, and other doctors in Chonburi hospital who performed tissue sampling and analysis and took responsibility for patient management. We also thank the cytotechnicians and laboratory workers in our department for skillful technical support.
REFERENCES
1) Trapnell BC, Whitsett JA, Nakata K. Pulmonary alveolar proteinosis. N Engl J Med. 2003; 349: 2527-2539.
2) Rosen SH, Castleman B, Liebow AA. Pulmonary alveolar proteinosis. N Engl J Med. 1958; 258: 1123-1142.
3) Doyle AP, Balcerzak SP, Wells CL, Crittenden JO. Pulmonary alveolar proteinosis with hematologic disorders. Arch Intern Med. 1963; 112: 940-946.
4) Ohnishi T, Yamada G, Shijubo N et al. Secondary Pulmonary alveolar proteinosis associated with myelodysplastic syndrome. Internal Medicine. 2003; 42: 187-190.
5) Shah PL, Hansell D, Lawson PR et al. Pulmonary alveolar proteinosis: clinical aspects and current concepts on pathogenesis. Thorax. 2000; 55: 67-77.
6) Chou CW, Lin FC, Tung SM et al. Diagnosis of pulmonary alveolar proteinosis: usefulness of Papanicolaou-stained smears of bronchoal-veolar lavage fluid. Arch Intern Med. 2001; 161: 562-566.
7) Maygarden SJ, Iacocca MV, Funkhouser WK, Novotny DB. Pulmonary alveolar proteinosis: a spectrum of cytologic, histochemical, and ultrastructural findings in bronchoalveolar lavage fluid. Diagn Cytopathol. 2001; 24: 389395.
8) Byun MK, Kim DS, Kim YW et al. Clinical features and outcomes of idiopathic pulmonary alveolar proteinosis in Korean population. J Korean Med Sci. 2010; 25: 393-398.
9) Xu Z, Jing J, Wang H et al. Pulmonary alveolar proteinosis in China: a systematic review of 241 cases. Respirology. 2009; 14: 761-766.
10) Sanders M, Kahan M, Sbar S. Pulmonary alveolar proteinosis: a review of the literature: report of two cases. Dis Chest. 1962; 42: 437441.
11) Gupta RK, Wakefield St J, Naran S et al. Cytohistologic and electron microscopic findings in bronchoalveolar lavage fluid in a case of pulmonary alveolar proteinosis. Diagn Cytopathol. 2002; 27: 63-65.
12) Bonella F, Bauer PC, Griese M et al. Pulmonary alveolar proteinosis: new insights from a single-center cohort of 70 patients. Respir Med. 2011; 105: 1908-1916.
13) Chotivanich K, Udomsangpetch R, Pattana-panyasat K et al. Hemoglobin E: a balanced polymorphism protective against high parasitemias and thus sever P falciparum malaria. Blood. 2002; 100: 1172-1176.
14) Vichinsky Elliott. Hemoglobin E syndromes. Hematology Am Soc Hematol Educ Program. 2007: 79-83.