

The detection of HLA-B27 allele types by quantitative real-time polymerase chain reaction with melting curve analysis
Supichaya Nimnuan-ngam1, Pasra Arnutti2* and Thirayost Nimmanon2
1 Siriraj Center of Research of Excellence in Immunoregulation, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok, Thailand
2 Department of Pathology, Phramongkutklao College of Medicine, Bangkok, Thailand
Correspondence to:
Colonel Assistant Professor Dr Pasra Arnutti,
Department of Pathology, Floor 6, Her Royal Highness Princess Bejaratana Building,
Phramongkutklao College of Medicine, 317 Rajavithi Road, Rajadevi, Bangkok 10400 Thailand.
Telephone: +66 (0) 83 619 8689 Fax: +66 (0) 2 354 7791 Email: pasra@pcm.ac.th, pasra@pcmpathology.org, pasra@hotmail.com
Conflict of interest: The authors declare that they have no conflicts of interest with the contents of this article.
Abstract
Human leucocyte antigen B27 (HLA-B27) is a major histocompatibility complex class 1 molecule that is strongly associated with ankylosing spondylitis. The polymerase chain reaction with sequence specific primers (PCR-SSP) is currently used for the discrimination of HLA-B27 allele types resulting from single base substitutions. However, this technique is a multi-step, time-consuming task and increases the chance of post-PCR contamination. The main objective of this study is to determine the HLA-B27 genotypes using quantitative real-time polymerase chain reaction (Q-PCR) followed by melting curve analysis. Genomic DNA (gDNA) was isolated from 50 outdated peripheral blood samples stored at -20 °C in the Molecular Genetics Laboratory, Department of Pathology, Phramongkutklao College of Medicine, Bangkok, Thailand. These blood specimens were previously collected from 25 patients each who were HLA-B27 negative and positive in PCR-SSP. The genotyping of HLA-B27 was detected in the isolated gDNA by a Q-PCR assay with melting curve analysis. All HLA-B27-positive blood samples yielded either a single peak or double peaks at 87.28 ± 1.36 °C. Only one peak for an endogenous reference gene (human beta-globin gene) was detectable at 84.90 ± 0.09 °C in all HLA-B27-negative blood samples. The Q-PCR followed by melting curve analysis is reliable for the DNA typing of the HLA-B27 alleles and could be used to an alternative to a conventional PCR-SSP method.
Keywords: human leucocyte antigen B27; melting curve analysis; quantitative real-time polymerase chain reaction
Introduction
Ankylosing spondylitis (AS) is a form of arthritis that effects the spine(1). It causes inflammation of the spinal joints that can lead to severe and chronic pain. Human leukocyte antigen B27 (HLA-B27) is associated with AS, including ocular diseases, rheumatic diseases and inflammatory bowel disease. Approximately 95% of patients with AS have HLA-B27 compared with only 8% of healthy individuals. Several methods have been developed for the identification of the HLA-B27 allele(1), i.e. serology, polymerase chain reaction (PCR), including the standard PCR with sequence-specific primers (SSP). However, PCR is a time-consuming process and also requires post-PCR manual procedures(2). Currently, the application of real-time PCR or quantitative PCR (qPCR) with a double-stranded DNA binding SYBR Green dye has facilitated the rapid detection and amplification of PCR products(3,4). The aim of this study was to develop of the qPCR analysis for HLA-B27 allele test in routine work.
Materials and Methods
Samples:
Fifty outdated peripheral blood samples were enrolled in this study. They are comprised of 25 HLA-B27-negative and 25 HLA-B27-positive blood samples. All blood samples were kept at -20 °C until DNA isolation and analysis(5). Genomic DNA was isolated from 500 µL of whole blood using the AxyPrep™ Blood Genomic DNA Miniprep Kit (CA, USA) according to the manufacturer's instructions.
Real-time PCR analysis:
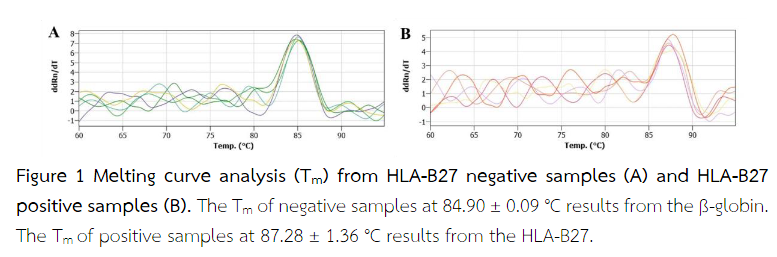
The real-time PCR was performed on qTower3-Real-Time PCR thermocycler (Analytik Jena AG, Germany). A total volume of 12 µL of each mixture consisted of 5 µl of SYBR Green I (EXPRESS SYBR™ GreenER™ qPCR Supermix, universal; Invitrogen, USA); 4 µL of detection mix containing the primer E91S and 136AS specific for HLA-B27, primer BG1 and BG2 specific for ß-globin(3); 1 µL of distilled water; and 2 µl of DNA template. The real-time PCR amplification protocol for this reaction consisted of an initial denaturation step at 95 °C for 5 sec, and 45 cycles of 95 °C for 4 sec, 68 °C for 1 min, 95 °C for 45 sec, 58 °C for 1 sec and final extension at 72 °C for 33 sec. After amplification was complete, a final melting curve analysis (Tm) was followed by the generation of a thermal gradient from 60 °C to 95 °C with a ramp rate of 5 °C/s. HLA-B27 positive samples give a unique melting peak at 87.28 ± 1.36 °C. Therefore, HLA-B27 negative samples showed a single temperature curve at 84.90 ± 0.09 °C representing the ß-globin(5,6).
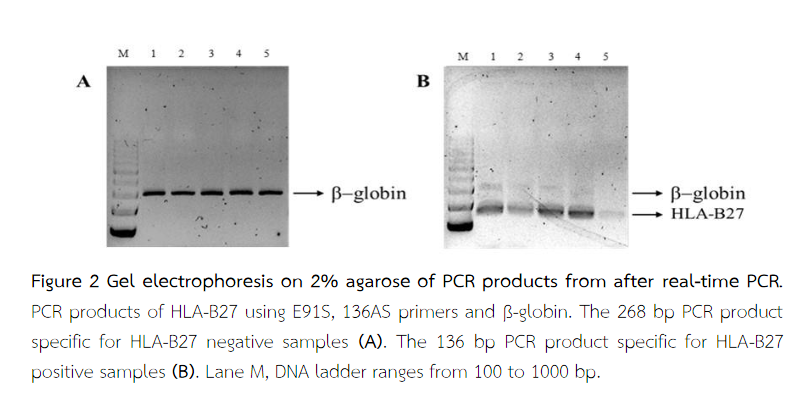
The results were confirmed on the agarose gel. The melting peak for ß-globin showed 268 bp PCR product and the melting peaks for HLA-B27 alleles showed 136 bp PCR product representative of HLA-B27-positive samples(5).
Results
The results of real-time PCR showed that among 50 samples, 25 samples were HLA-B27-positive and 25 samples were HLA-B27-negative. All negative samples had only one peak at 84.90 ± 0.09 °C (Figure 1A). All positive samples showed single or double peaks at 87.28 ± 1.36 °C (Figure 1B). This signal was absent for the HLA-B27 negative samples. The melting peaks for ß-globin (Tm = 84.90 ± 0.09 °C) and the 268 bp PCR product were visible on the agarose gel correlated with HLA-B27-negative samples (Figure 2A). The melting peaks for HLA-B27 (Tm = 87.28 ± 1.36 °C) and the 136 bp PCR product are visible on the agarose gel correlated with HLA-B27-positive samples (Figure 2B). Despite the lower fragment length, the HLA-B27 specific PCR product (136 bp) yielded a higher Tm value compared with the ß-globin 268 bp product because of a higher GC content(5).
Discussions
We compared the results of 50 genotyped individual with our previous original PCR method. All genotypes were completely concordant. However, conventional PCR requires post-PCR manipulations that increase the risk of cross-contamination between samples. These post-PCR steps are laborious, especially when large numbers of samples and cost-effective(6,7). Thus, we are starting point for development of a real-time PCR. Real-time PCR is one of the methods used to detect for HLA-B27 allele in patients suspected to have AS and related diseases. Real-time PCR is much more accurate than PCR. Real-time PCR substantially reduced the labor-intensive steps and the total processing time when compared to the usual time required for PCR. It is therefore a suitable in routine laboratory practice(8).
Conclusion
The real-time PCR assay is possibly reliable for the detection of HLA-B27.
References
- Eun Hae Cho MD, Sang Gon Lee MD, Jeong Ho Seok MD, Bo Ya Na Park MS, Eun Hee Lee MD. Evaluation of Two Commercial HLA-B27 Real-Time PCR Kits. Korean J Lab Med. 2009;29:589-93.
- Nätterkvist Y. Development of a PCR method to detect HLA-B27 in ankylosing spondylitis. Department of Medical Biochemistry and Microbiology. 2012;15:1-22.
- Tiemann C, Vogel A, Dufaux B, Zimmer M, Krone J-R, Hagedorn H-J. Rapid DNA Typing of HLA-B27 Allele By Real-Time PCR Using LightCycler Technology. Clin Lab. 2001;47:131-4.
- Gersuk VH, Nepom GT. A real-time PCR approach for rapid high resolution subtyping of HLA-DRB1*04. J Immunol Methods. 2007;317:1-11.
- Bon MAM, Oeveren-Dybicz Av, Bergh FAJTMvd. Genotyping of HLA-B27 by Real-Time PCR without Hybridization Probes. Clinical Chemistry. 2000;7:1000-2.
- Faner R, Casamitjana N, Colobran R, Ribera A, Pujol-Borrell R, Palou E, et al. HLA-B27 Genotyping by Fluorescent Resonance Emission Transfer (FRET) Probes in Real-Time PCR. Human Immunology. 2004;65:826-38.
- Panichrum P, Chuesakul M, Fungkajai M, Kaset C, Nathalang O. HLA-B27 Screening Using PCR-SSP Technique in Thai Blood Donors. J Med Tech Assoc Thailand. 2013;41:4440-5.
- Casamitjana N, Faner R, Santamaria A, Colobran R, Ribera A, Pujol-Borrell R, et al. Development of a New HLA-DRB Real-Time PCR Typing Method. Human Immunology. 2005;66:85-91.